
User:Tanganu/sandbox

The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen.[1] Hence, the reaction is sometimes referred to as the Huisgen cycloaddition, although this term is often used to specifically describe the 1,3-dipolar cycloaddition between an organic azide and an alkyne to generate 1,2,3-triazole. Currently, 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives.

Mechanistic Overview
[edit]There were two original proposals for the mechanism of the 1,3-dipolar cycloaddition: the concerted pericyclic cycloaddition mechanism, proposed by Rolf Huisgen,[2] and the stepwise mechanism involving a diradical intermediate, proposed by Firestone.[3] The former proposal is now generally accepted.[4] The 1,3-dipole reacts with the dipolarophile in a concerted, often asynchronous, and symmetry-allowed π4s + π2s fashion through a six-electron Huckel aromatic transition state.
Pericyclic Mechanism
[edit]Huisgen's proposal was supported by a series of experiments on the reaction between the 1,3-dipolar diazo compounds and various dipolarophilic alkenes, as outlined in the figure below.[5] Theoretically, this reaction can proceed in either a concerted pericyclic fashion or a stepwise manner. If the reaction proceeded through the stepwise mechanism, it would involve the formation of a charge-separated intermediate. However, the following observations refuted the existence of such an intermediate, and hence supported the pericyclic mechanism:
- Substituent effect: For the stepwise mechanism, there is a negative charge build-up on the dipole in the intermediate. Electron-withdrawing substituents at this position should stabilize this intermediate and therefore speed up the reaction. However, electrion-withdrawing groups actually lower the reactivity, suggesting that this reaction is likely not stepwise.
- Solvent effect: The stepwise mechanism involves a highly polar (charge-separated) transition state. Generally, such a reaction proceeds faster in a polar solvent, in which the polar transition state can be stabilized. However, solvent polarity exerts little effect on the rate of 1,3-dipolar cycloadditions, disputing the stepwise mechanism.
- Stereochemistry: 1,3-dipolar cycloadditions are always stereospecific with respect to the substituents on the dipolarophile (i.e., cis-alkenes giving syn-products), supporting the concerted pericyclic mechanism where both sigma bonds are formed simultaneously. If the reaction were stepwise, bond rotation in the intermediate would be possible and the stereochemistry would get scrambled.
- Thermodynamic parameters: Bimolecular reactions require associative collision between two reactants, so they have negative entropies of activation (ΔS‡ < -14 e.u.). 1,3-dipolar cycloadditions, however, have an unusually large negative entropy of activation (ΔS‡ ~ -30 e.u.) similar to that of the Diels-Alder reaction (ΔS‡ ~ -30 to -40 e.u.), suggesting that the transition state is highly ordered, which is a signature of concerted pericyclic reactions.

1,3-Dipole
[edit]A 1,3-dipole is a chemical species that can be represented as either an allyl-type or a propargyl/allenyl-type zwitterionic octet/sextet structures. Both types of 1,3-dipoles share four electrons in the π-system over three atoms. The allyl-type is bent whereas the propargyl/allenyl-type is linear in geometry. There are a total of 12 allyl-type and 6 propagyl/allenyl-type second-row 1,3-dipoles (which consist of carbon, nitrogen and oxygen centers). All of these dipoles are listed and named in the table below.[6] 1,3-Dipoles containing higher-row elements such as sulfur or phosphorus are also known, but are utilized less routinely.

For any given 1,3-dipole, resonance structures can be drawn such that the negative charge and the positive charge is delocalized on either of the two termini (see the resonance scheme below). Such ambivalence makes it difficult to distinguish which terminus is electron-rich and which is electron-poor based on visual inspection alone. A more accurate method to describe the electronic distribution on a 1,3-dipole is to assign the major resonance contributor based on experimental or theoretical data. For example, both dipole moment measurement[7] and computation[8] of diazomethane agreeably assign the highest negative character to the terminal nitrogen atom. In contrast, hydrazoic acid has the largest negative character at the internal nitrogen atom (see figure below). The electronic distribution contributes to the polarity of the 1,3-dipole -- if the charge is distributed over the two termini evenly, then the 1,3-dipole will have little polarity.

Consequently, this ambivalence means that the termini of a 1,3-dipole can be treated as both nucleophilic and [[electrophile|electrophilic] at the same time. The extent of nucleophilicity and electrophilicity at each terminus can be assigned more rigorously by the frontier molecular orbitals of the dipole, which can be obtained computationally. In general, the center carrying the biggest lobe (i.e., largest orbital coefficient) in the HOMO is the most nucleophilic site, whereas the center bearing biggest lobe in the LUMO is the most electrophilic site. The most nucleophilic site is generally also the most electron-rich site. However, it is important to note that this is not always the case, as orbital lobe size does not necessarily correspond to the degree of charge carried by that orbital. For example, calculation shows that diazomethane bears the highest negative charge on the terminal nitrogen atom, yet it reacts as an electrophile at this terminal nitrogen atom and as a nucleophile or a base at the carbon atom (see scheme below) [9] [10] [11] Depending on the identity of the dipole-dipolarophile pair, the nucleophilic or the electrophilic character of the 1,3-dipole may dominate. For example, ozone acts prominently as an electrophilic dipole, whereas diazo compounds act primarily as electron-rich nucleophilic dipoles. Some other dipoles, such as organic azides, can behave as an electrophile or a nucleophile depending on their dipolarophilic partner.

Dipolarophile
[edit]The most commonly used dipolarophiles are alkenes and alkynes, but heteroatom-containing dipolarophiles such as carbonyls and imines can also undergo 1,3-dipolar cycloaddition. Other examples of dipolarophiles include fullerenes and nanotubes, which can undergo 1,3-dipolar cycloaddition with azomethine ylide in the Prato reaction.
Solvent Effect
[edit]In polar reactions, polar solvents strongly stabilize the polar transition states relative to the non-polar reactants. Thus, polar solvents accelerate the rate of polar reactions where charge separation is observed in the transition state. However, 1,3-dipolar cycloadditions experience very little solvent effect, strongly suggesting that they are concerted pericyclic reactions with little polarity change from reactants to the transition state rather than stepwise reactions. The rate of reaction between phenyl diazomethane and ethyl acrylate or norbornene (see scheme below) increases only 2- to 6-fold, respectively, in non-polar cyclohexane to highly polar methanol as solvents.[12]

A clear demonstration of lack of solvent effect in 1,3-dipolar cycloaddition is the reaction between enamines and dimethyl diazomalonate (see scheme below).[13] N-cyclopentenyl pyrrolidine does not undergo cycloaddition with dimethyl diazomalonate. Rather, the enamine adds nucleophilically to the terminal nitrogen atom of the diazo group, generating a charge-separated intermediate. Subsequent proton exchange gives the ultimate product. This reaction proceeds 1,500 times faster in polar DMSO than in nonpolar decalin. On the other hand, a close analog, N-cyclohexenyl pyrrolidine adds to dimethyl diazomalonate in the 1,3-dipolar cycloaddition manner to yield the five-membered ring product. This reaction is sped up only 40-fold in DMSO relative to decalin. Such disparity in solvent effects in the nucleophilic addition versus cycloaddition strongly suggests the cycloaddition does not proceed through a charge-separated intermediate.

Frontier molecular orbital theory
[edit]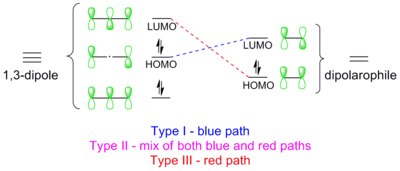
1,3-Dipolar cycloadditions are thermally allowed, π4s + π2s reactions. They can be treated equivalently by the Woodward-Hoffmann rules and the Dewar-Zimmerman theory. In the Dewar-Zimmerman treatment, 1,3-dipolar cycloadditions proceed through a 5-center, zero-node, 6-electron Huckel transition state, which is thermally allowed. In the Woodward-Hoffmann treatment, frontier molecular orbitals (FMO) in the 1,3-dipole must overlap with corresponding FMOs of the same symmetry in the dipolarophile. According to the FMO diagram drawn above, such an symmetry-allowed orbital overlap can be achieved in three ways, which are termed types I, II and III,[14] the explanations of which are given in the following section. Whichever FMO pair has the smallest energy gap will dominate the reaction.

Type I
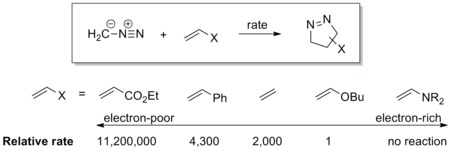
[edit]The dipole has a high-lying HOMO which overlaps with LUMO of the dipolarophile (indicated by blue dashed lines in the diagram). A dipole of this class is referred to as a HOMO-controlled dipole or a nucleophilic dipole, which includes azomethine ylide, carbonyl ylide, nitrile ylide, azomethine imine, carbonyl imine and diazoalkane. These dipoles add to electrophilic alkenes readily. Electron-withdrawing groups (EWG) on the dipolarophile would accelerate the reaction by lowering the LUMO, while electron-donating groups (EDG) would decelerate the reaction by raising the HOMO. For example, the reactivity scale of diazomethane against a series of dipolarophiles is shown in the scheme below. Diazomethane reacts with the electron-poor ethyl acrylate more than a million times faster than the electron rich butyl vinyl ether.[15]
Type II
[edit]HOMO of the dipole can pair with LUMO of the dipolarophile; alternatively, HOMO of the dipolarophile can pair with LUMO of the dipole. This two-way interaction arises because the energy gap in either direction is similar. A dipole of this class is referred to as a HOMO-LUMO-controlled dipole or an ambiphilic dipole, which includes nitrile imine, nitrone, carbonyl oxide, nitrile oxide, and azide. Any substituent on the dipolarophile would accelerate the reaction by lowering the energy gap between the two interacting orbitals; i.e., an EWG would lower the LUMO while an EDG would raise the HOMO. For example, azides react with various electron-rich and electron-poor dipolarophile with similar reactivities (see reactivity scale below).[16]

Type III
[edit]The dipole has a low-lying LOMO which overlaps with HOMO of the dipolarophile (indicated by red dashed lines in the diagram). A dipole of this class is referred to as a LUMO-controlled dipole or an electrophilic dipole, which includes nitrous oxide and ozone. EWGs on the dipolarophile decelerate the reaction, while EDGs accelerate the reaction. For example, ozone reacts with the electron-rich 2-methylpropene about 100,000 times faster than the electron-poor tetrachloroethene (see reactivity scale below).[17]

Reactivity
[edit]Concerted processes such as the 1,3-cycloaddition require a highly ordered transition state (high negative entropic energy) and only moderate enthalpy requirements. Using competition reaction experiments, relative rates of addition for different cycloaddition reactions have been found to offer general findings on factors in reactivity.
- Conjugation, especially with aromatic groups, increases the rate of reaction by stabilization of the transition state. During the transition, the two sigma bonds are being formed at different rates, which may generate partial charges in the transition state that can be stabilized by charge distribution into conjugated substituents.
- More polarizable dipolarophiles are more reactive because diffuse electron clouds are better suited to initiate the flow of electrons.
- Dipolarophiles with high angular strain are more reactive due to increased energy of the ground state.
- Increased steric hindrance in the transition state as a result of unhindered reactants dramatically lowers the reaction rate.
- Hetero-dipolarophiles add more slowly, if at all, compared to C,C-diapolarophiles due to a lower gain in sigma bond energy to offset the loss of a pi bond during the transition state.
- Isomerism of the dipolarophile affects reaction rate due to sterics. trans-isomers are more reactive (trans-stilbene will add diphenyl(nitrile imine) 27 times faster than cis-stilbene) because during the reaction, the 120° bond angle shrinks to 109°, bringing eclipsing cis-substituents towards each other for increased steric clash.

See Huisgen reference doi:10.1002/anie.196306331.

Stereospecificity
[edit]1,3-dipolar cycloadditions always result in retention of stereochemistry with respect to both the 1,3-dipole and the dipolarophile. Such high degree of stereospecificity is a strong support for the concerted over the stepwise reaction mechanism. Regarding the dipolarophile, determination of stereospecificity is simple: cis-substituents on the dipolarophilic alkene end up syn, and trans-substituents end up anti in the five-membered ring product (see scheme below).[18]

Retention of the 1,3-dipole stereochemistry is more complex to study. Out of the 18 possible second-row 1,3-dipoles shown above, only two species that possess two stereogenic centers are relevant to the determination of stereospecificity: azomethine ylide and carbonyl ylide. The stereochemistry of these molecules are challenging to control because rotation about the single bond would scramble the stereochemistry of the 1,3-dipole reagent. Nonetheless, it is possible to generate these 1,3-dipoles in a stereocontrolled manner through cycloreversion of aziridines, and then rapidly trap them with dipolarophiles before rotation about the single bond can take place. The scheme below[19] shows the thermal or photochemical conversion of aziridines into azomethine ylide with the desired stereochemistry. Trapping of these dipoles with strong dipolarophile indeed reveals the retention of stereochemistry of the dipole. However, non-stereospecific products are observed when weaker electrophiles are utilized as the trap; the slow cycloaddition allows rotation about the single bond and thus stereochemical scrambling to take place (see scheme below). Consequently, the reaction is stereospecific with respect to both the 1,3-dipole and the dipolarophile, giving retention to both of them in the same reaction. Generation of stereospecific carbonyl ylide can also be achieved by cycloreversion of epoxides.[20]

Lastly, the stereochemistry of the central atom of a 1,3-dipole has been found to follow the principle of least motion; i.e., the position of this substituent on the central atom does not change much going from the starting material to the product. For example, in the cycloaddition of nitronic esters (N-alkoxy nitrones) to alkenes, the central-atom alkoxy group ends up axial in the envelope of the five-membered ring product as predicted by least motion (see scheme below).[21]

Diastereoselectivity
[edit]When two or more chiral centers are generated during the cycloaddition reaction, diastereomeric transition states and products can be obtained. There are two forces that influence the diastereoselectivity of 1,3-dipolar cycloadditions: the attractive π-interaction (resembling secondary orbital interactions in the Diels-Alder cycloaddition) and the repulsive steric interaction. Often these two forces can offset each other and give poor diastereoselection. An example of a diastereoselective 1,3-dipolar cycloaddition is illustrated by the reaction between benzonitrile N-benzylide and methyl acrylate. In the transition state, the phenyl and the methyl ester groups interact favorably, giving the cis-substitution as the exclusive final pyroline product. This favorable π-interaction offsets the steric repulsion between the phenyl and the methyl ester groups.[22]

Regioselectivity
[edit]For asymmetric dipole-dipolarophile pairs, two regioisomeric products are possible. Both electronic/stereoelectronic and steric factors contribute to the regioselectivity of 1,3-dipolar cycloadditions. With respect to the electronic/stereoelectronic consideration, the interaction between two orbitals with the largest orbital coefficients (i.e., the largest HOMO orbital with the largest LUMO orbital) is the most dominant interaction. Depending on the type of the 1,3-dipolar cycloaddition (type I, II or III), one can often computationally determine the largest HOMO and LUMO combinations and predict the regiochemical outcome of the reaction.[23][24] For example, the scheme below illustrates the cycloaddition of diazomethane to three dipolarophiles: methyl acrylate, styrene or methyl cinnamate. In all three cases, diazomethane acts as a nucleophilic dipole, with the largest HOMO lobe residing on the carbon atom, and the dipolarophiles are electrophilic species. Methyl acrylate and styrene bear the largest LUMO lobe on the terminal olefinic carbon -- this can be easily predicted based on resonance structures. Hence, the regiochemistry of the cycloaddition should be dominated by the interaction between the carbon atom of diazomethane and the terminal olefinic carbon of the dipolarophile, giving the carboxyl or the phenyl substitution at the C-3 position. For the more complex case of methyl cinnamate, the two substituents compete at withdrawing electrons from the alkene. Nonetheless, the carboxyl is more electron-withdrawing, making the beta-carbon the more electrophilic center of this molecule. The cycloaddition should yield the carboxyl group on C-3 and the phenyl group on C-4 in the cyclic product. It is important to note that while this approach works in many cases, it is not always applicable.

The second important factor that influences regiochemistry is steric effect. Steric effects can cooperate or compete with the aforementioned electronic orbital overlap and can sometimes completely override the electronic preference, giving the opposite regioisomer. For example, electronic treatment of the cycloaddition between diazomethane and methyl acrylate yield the 3-carboxyl pyrazoline as the major product, as illustrated by the diagram above. However, the more sterically demanding 2-diazopropane reacts with a series of methyl acrylate derivatives to yield a mixture of 3- and 4-carboxyl pyrazoline, as outlined in the scheme below. The ratio of these two products depends on the size of the substituent on methyl acrylate. Increasing the size from hydrogen to t-butyl shifts the regioselectivity from 100% 3-carboxyl to 100% 4-carboxyl substitution. The steric effect is even more pronounced using propiolate derivatives, as the electronic preference is smaller in propiolates than in acrylates.[25][26]

Product orientation
[edit]Although there are general guidelines for orientation of products, the actual selectivity in these cycloaddition reactions is varied. For example, in azide-alkyne coupling reactions without Cu(I) catalyst, the products are generated in a 1:1 ratio of 1,4 and 1,5 products.[27]
In reactions with hetero-dipolarophiles, the direction of addition is based on the maximization of gains in sigma bond energy during reaction. However, there are several exceptions that include multistep reaction pathways.
When the dipolarophiles are alkenes or alkynes, the dominant force in dictating direction of addition are substituents effects, primarily steric.[28]

Synthetic Applications
[edit]Nitrile oxides
[edit]Nitrile oxides are commonly prepared from the corresponding aldehydes through oxime intermediates, or from the nitro precursor. Stereospecific cycloaddition between a nitrile oxide and an alkene yields the cyclic isoxazoline product, whereas the reaction with an alkyne yields the isoxazole instead. Both isoxazolines and isoxazoles are of tremendous synthetic use because they can be cleaved by hydrogenation to reveal aldol-type β-hydroxycarbonyl or Claisen-type β-dicarbonyl products, respectively.

Nitrile oxide-alkyne cycloaddition followed by hydrogenation was utilized in the synthesis of Miyakolide as illustrated in the figure below.[29]

Carbonyl ylides
[edit]Carbonyl ylides can be synthesized by trapping carbenes with carbonyl oxygens. For example, treatment of α-diazocompounds with Rh2(OAc)4 makes an α-carbene, which can be intercepted by a carbonyl group in situ to generate the carbonyl ylide.

1,3-dipolar cycloaddition between a carbonyl ylide and an alkenes yields a furan-type oxacycle. This reaction has been applied in the synthesis of challenging targets such as phorbol.[30]

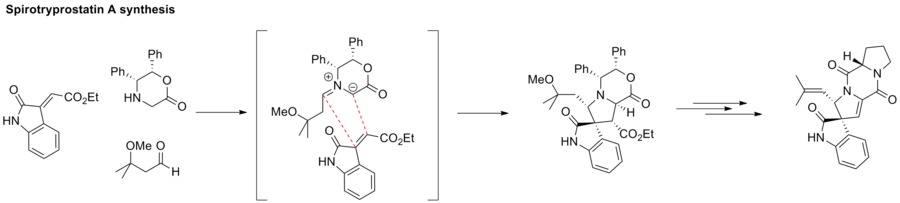
Azomethine ylides
[edit]1,3-Dipolar cycloaddition between an azomethine ylide and an alkene furnishes an azacyclic structure. This strategy has been applied to the synthesis of spirotryprostatin A.[31]
Biological Applications
[edit]Azide-Alkyne
[edit][32] ===
Lopez, S. A.; Houk, K. N. J. Org. Chem. 2012, ASAP.== References ==
- ^ Huisgen, Rolf (1963). "1.3-Dipolare Cycloadditionen Ruckschau und Ausblick" (abstract). Angewandte Chemie. 75 (13): 604–637. doi:10.1002/ange.19630751304.
- ^ Huisgen, Rolf (November 1963). "Kinetics and Mechanism of 1,3-Dipolar Cycloadditions". Angewandte Chemie International Edition. 2 (11): 633–645. doi:10.1002/anie.196306331.
- ^ Fireston, R (1968). "Mechanism of 1,3-dipolar cycloadditions". Journal of Organic Chemistry. 33 (6): 2285–2290. doi:10.1021/jo01270a023.
- ^ Huisgen, Rolf (1976). "1,3-Dipolar cycloadditions. 76. Concerted nature of 1,3-dipolar cycloadditions and the question of diradical intermediates". Journal of Organic Chemistry. 41 (3): 403–419. doi:10.1021/jo00865a001.
- ^ Huisgen, Rolf (November 1963). "Kinetics and Mechanism of 1,3-Dipolar Cycloadditions". Angewandte Chemie International Edition. 2 (11): 633–645. doi:10.1002/anie.196306331.
- ^ Huisgen, Rolf (1963). "1,3-Dipolar Cycloadditions. Past and Future". Angewandte Chemie International Edition. 2 (10): 565–598. doi:10.1002/anie.196305651.
- ^ Cox, A; Thomas, L; Sheridan, J (1958). "Microwave Spectra of Diazomethane and its Deutero Derivatives". Nature. 181 (4614): 1000–1001. doi:10.1038/1811000a0.
- ^ Hilberty, P; Leforestier, C (1978). "Expansion of molecular orbital wave functions into valence bond wave functions. A simplified procedure". Journal of the American Chemical Society. 100 (7): 2012–2017. doi:10.1021/ja00475a007.
- ^ McGarrity, J.F.; Patai, Saul (1978). "Basicity, acidity and hydrogen bonding". Diazonium and Diazo Groups: Volume 1 date = 1978 url = http://onlinelibrary.wiley.com/doi/10.1002/9780470771549.ch6/summary: 179–230. doi:10.1002/9780470771549.ch6. ISBN 9780470771549.
{{cite journal}}: External link in|journal=|journal=(help) - ^ Berner, Daniel; McGarrity, John (1979). "Direct observation of the methyldiazonium ion in fluorosulfuric acid". Journal of the American Chemical Society. 101 (11): 3135–3136. doi:10.1021/ja00505a059.
- ^ Muller, Eugen; Rundel, Wolfgans (1956). "Untersuchungen an Diazomethanen, VI. Mitteil.: Umsetzung von Diazoäthan mit Methyllithium". Chemische Berichte. 89 (4): 1065–1071. doi:10.1002/cber.19560890436.
- ^ Geittner, Jochen; Huisgen, Rolph; Reissig, Hans-Ulrich (1978). "Solvent Dependence of Cycloaddition Rates of Phenyldiazomethane and Activation Parameters". Heterocycles. 11: 109–120. doi:10.3987/S(N)-1978-01-0109.
- ^ Huisgen, Rolph; Reissig, Hans-Ulrich; Huber, Helmut; Voss, Sabine (1979). "α-Diazocarbonyl compounds and enamines - a dichotomy of reaction paths". Tetrahedron Letters. 20 (32): 2987–2990. doi:10.1016/S0040-4039(00)70991-9.
- ^ Sustmann, R (1974). "Orbital energy control of cycloaddition reactivity". Pure and Applied Chemistry. 40 (4): 569–593. doi:10.1351/pac197440040569.
- ^ Geittner, Jochen; Huisgen, Rolf (1977). "Kinetics of 1,3-dipolar cycloaddition reactions of diazomethane; A correlation with homo-lumo energies". Tetrahedron Letters. 18 (10): 881–884. doi:10.1016/S0040-4039(01)92781-9.
- ^ Huisgen, Rolf; Szeimies, Gunter; Mobius, Leander (1967). "K1.3-Dipolare Cycloadditionen, XXXII. Kinetik der Additionen organischer Azide an CC-Mehrfachbindungen". Chemische Berichte. 100 (8): 2494–2507. doi:10.1002/cber.19671000806.
- ^ Williamson, D. G.; Cvetanovic, R. J. (1968). "Rates of ozone-olefin reactions in carbon tetrachloride solutions". Journal of the American Chemical Society. 90 (14): 3668–3672. doi:10.1021/ja01016a011.
- ^ Bihlmaier, Werner; Geittner, Jochen; Huisgen, Rolf; ReissigP, Hans-Ulrich (1978). "The Stereospecificity of Diazomethane Cycloadditions". Heterocycles. 10: 147–152. doi:10.3987/S-1978-01-0147.
- ^ Huisgen, Rolf; Scheer, Wolfgang; Huber, Helmut (1967). "Stereospecific Conversion of cis-trans Isomeric Aziridines to Open-Chain Azomethine Ylides". Journal of the American Chemical Society. 89 (7): 1753–1755. doi:10.1021/ja00983a052.
- ^ Dahmen, Alexander; Hamberger, Helmut; Huisgen, Rolf; Markowski, Volker (1971). "Conrotatory ring opening of cyanostilbene oxides to carbonyl ylides". Journal of the Chemical Society D: Chemical Communications (19): 1192–1194. doi:10.1039/C29710001192.
- ^ Gree, R.; Carrie, R. (1976). "Cycloadditions dipolaires-1,3-XXV: Formation sous controle cinetique d'invertomeres stables lors des cycloadditions d'esters nitroniques". Tetrahedron. 32: 683–688. doi:10.1016/S0040-4020(01)93791-3.
- ^ Padwa, Albert; Smolanoff, Joel (1971). "Photocycloaddition of arylazirenes with electron-deficient olefins". Journal of the American Chemical Society. 93 (2): 548–550. doi:10.1021/ja00731a056.
- ^ Caramella, Pierluigi; Houk, K.N. (1976). "Geometries of nitrilium betaines. The clarification of apparently anomalous reactions of 1,3-dipoles". Journal of the American Chemical Society. 98 (20): 6397–6399. doi:10.1021/ja00436a062.
- ^ Caramella, Pierluigi; Gandour, Ruth W.; Hall, Janet A.; Deville, Cynthia G.; Houk, K. N. (1977). "A derivation of the shapes and energies of the molecular orbitals of 1,3-dipoles. Geometry optimizations of these species by MINDO/2 and MINDO/3". Journal of the American Chemical Society. 99 (2): 385–392. doi:10.1021/ja00444a013.
- ^ Padwa, Albert (1983). 1,3-Dipolar Cycloaddition Chemistry. General Heterocyclic Chemistry Series. Vol. 1. United States of America: Wiley-Interscience. pp. 141–145. ISBN 0-471-08364-X.
- ^ Koszinowski, J. (1980). (Ph.D. Thesis).
{{cite thesis}}: Missing or empty|title=(help) - ^
Vsevolod V. Rostovtsev, Luke G. Green, Valery V. Fokin, K. Barry Sharpless (2002). "A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective Ligation of Azides and Terminal Alkynes". Angewandte Chemie International Edition. 41 (14): 2596–22599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. PMID 12203546.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ Huisgen, Rolf (November 1963). "Kinetics and Mechanism of 1,3-Dipolar Cycloadditions" (abstract). Angewandte Chemie International Edition. 2 (11): 633–645. doi:10.1002/anie.196306331.
- ^ Evans, David; Ripin, David; Halstead, David; Campos, Kevin (1999). "Synthesis and Absolute Stereochemical Assignment of (+)-Miyakolide". Journal of the American Chemical Society. 121 (29): 6816–6826. doi:10.1021/ja990789h.
- ^ Wender, Paul; Rice, Kenneth; Schnute, Mark (1997). "The First Formal Asymmetric Synthesis of Phorbol". Journal of the American Chemical Society. 119 (33): 7898. doi:10.1021/ja9706256.
- ^ Onishi, Tomoyuki; Sebahar, Paul; Williams, Robert (2003). "Concise, Asymmetric Total Synthesis of Spirotryprostatin A". Organic Letters. 5 (17): 3135–3137. doi:10.1021/ol0351910. PMID 12917000.
- ^ Lopez, Steven A (2012). Journal of Organic Chemistry. ASAP (ASAP). doi:10.1021.jo301267b (inactive 2023-08-02).
{{cite journal}}: Check|doi=value (help); Missing or empty|title=(help); Unknown parameter|coauthors=ignored (|author=suggested) (help)CS1 maint: DOI inactive as of August 2023 (link)