
User:Alejandro103/Sulfur isotope biogeochemistry
| This is the sandbox page where you will draft your initial Wikipedia contribution.
If you're starting a new article, you can develop it here until it's ready to go live. If you're working on improvements to an existing article, copy only one section at a time of the article to this sandbox to work on, and be sure to use an edit summary linking to the article you copied from. Do not copy over the entire article. You can find additional instructions here. Remember to save your work regularly using the "Publish page" button. (It just means 'save'; it will still be in the sandbox.) You can add bold formatting to your additions to differentiate them from existing content. |
Stable Isotopes in Plants
[edit]Methods of detection
[edit]Previous efforts to understand how sulfur metabolism and biosynthetic pathways relied on expensive labeling experiments using radioactive 35S. By leveraging natural assimilatory processes, stable isotopes ratios can be used to track what are the sources of sulfur for plants, what organs plants utilize in sulfur acquisition and how sulfur moves through plants.

Sulfur (S) stable isotope composition measurements are often done using an Elemental Analysis-Isotope Ratio Mass Spectrometer, (EA-IRMS) [1] in which organic sulfur from biological samples is oxidized to sulfur dioxide (SO2) and analysed on a mass spectrometer. The gas is then analyzed for the ratio of the lighter (32S16O2) to the heavier (34S16O2) isotopologue and this ratio is then compared to sulfur isotope standards in order to calculate relative fractionations. In biological materials, sulfur is particularly scarce, making the abundance of S isotopes difficult to measure. The elemental S composition of plant matter is ≈0.2%, accounting for approximately 2 mmol/m2 in most leaf tissue[2]. In order to reach detectable levels of 30 ng to 3 µg of elemental S to calculate reliable δ34S values, leaf tissue samples need to be between 2-5 mg.
Improvements in detection have been made in recent years in the utilization of gas chromatography coupled with multicollector ICP-MS (GC/MC-ICP-MS)[3] to be able to measure pmol quantities of organic S.Additionally, ICP-MS has been used to measure nanomolar quantities of dissolved sulfate[4]. Most studies have focused on measuring the bulk δ34S of plant tissues and few studies have been performed on measuring the δ34S of individual S-containing compounds. The coupling of high-performance liquid chromatography (HPLC) with ICP-MS has been proposed as a way to test individual S-containing compounds[2].
Sources
[edit]Each year, approximately 0.3 gigatons of elemental sulfur is converted into organic matter by photosynthetic organisms[5]. This organic sulfur is allocated into a diversity of compounds such as amino acids – namely cysteine (Cys) and methionine (Met) – proteins, cofactors, antioxidants, sulfate groups, Fe-S centers and secondary metabolites. The three main sources of sulfur are atmospheric, soil, and aquatic.
Most vegetation can acquire sulfur from gaseous atmospheric compounds or various ions either in soil solutions or water bodies[6]. Uptake of gaseous and dissolved sulfur compounds apparently occurs with little accompanying isotopic selectivity [7]. Dissolved sulfate (SO42-) is considered to be the central pool which is metabolized by microorganisms and plants as most forms of atmospheric sulfur is oxidized into sulfate. Atmospheric sulfur is eventually returned to the soil when it is scrubbed from the atmosphere during precipitation or through dryfall[6].
Atmosphere
[edit]Many plants acquire sulfur through gaseous atmospheric compounds. Leaves of trees have δ34S values lying between those of air and soil, suggesting that there is uptake occurring from atmospheric and soil sources. The δ34S values of trees has also been demonstrated to be height dependent with the foliage at the tops of conifers, bull rushes and deciduous trees having δ34S values more reflective of the atmosphere and lower foliage having δ34S values closer to that of soil [6]. It has been proposed that this is due to upper foliage exerting a canopy action on the lower branches, taking up atmospheric sulfur before it can reach lower levels. This is further supported with the epiphytic lichens and mosses having δ34S values close to atmospheric S compounds. This occurs due lichen and mosses having no access to soil and relying on the direct uptake of gaseous sulfur, dissolved sulfur through rainfall and dry fall accumulation, providing a cumulative record of atmospheric sulfur isotope composition.[6][8]
Main forms of atmospheric sulfur come from the natural sulfur emissions formed biologically and emitted as H2S or organic sulfur gases such as DMS (dimethyl sulfide), COS (carbonyl sulfide), and CS2 (carbon disulfide). These gases are predominantly formed over oceans, wetlands, salt marshes, and estuaries by algae and bacteria [9]. Anthropogenic emissions have increased the concentration of sulfur in the atmosphere mainly through emissions of SO2, from coal, oil, industrial processes, and biomass burning. In 2000, global anthropogenic emission of sulfur was estimated of 55.2-68 Tg S per year, which is much higher than the natural sulfur emissions estimated to be 34 Tg S per year [9]. In the event of excess sulfur in plant tissue it has been demonstrated that when exposed to high doses of sulfur dioxide, plants emit hydrogen sulfide (H2S) and possibly other reduced sulfur compounds in response to high sulfur loading[7]
Soil
[edit]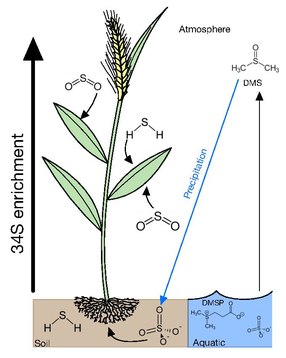
If soil sulfur is derived consistently from one source, the water-soluble and insoluble organic S fractions acquire similar isotopic compositions. In the case that there are two or more sources and/or if the isotopic composition of atmospheric or groundwater sulfate fluctuates, there may not be sufficient time for isotopic homogenization among the various forms of sulfur. The primary form of sulfur in soil is sulfate, which is transported upwards through the root system with minimal δ34S fractionation by 1-2‰[10]. In contrast to higher canopy plants reflecting atmospheric δ34S, protected understory plants tend to reflect soil sulfur[6].
Aquatic
[edit]The forms of sulfur available in aquatic environments depends on whether it is a marine or freshwater environment. In marine environments, the main forms of sulfur available is in sulfate at ~29mM and a δ34S of 21‰ in the surface. This excess in sulfur is subsequently converted into dimethylsulfoniopropionate (DMSP) by algae as an osmolyte and a repellent against grazing. DMSP also accounts for 50-100% of bacterial sulfur demand making it the most important source of reduced sulfur for marine bacteria. DMSPs cleavage product dimethyl sulfide (DMS) is highly volatile escaping the ocean into the atmosphere with emissions ranging between 15 and 33 Tg S year–1 [9] and accounting for 50-60% of the total natural reduced sulfur flux to the atmosphere.
Freshwater environments are more varied and depend on a multitude of factors, such as atmospheric deposition, runoff, diagenesis of bedrock and the presence of microbial sulfate reducers (MSR). Overall, the main sources of sulfur in freshwater environments are hydrogen sulfide and sulfate. In estuaries, plant roots extend into sulfide rich δ34S depleted sediments, created by MSR, and incorporate that into their biomass. Though levels of sulfide produced by MSR can be toxic and it has been proposed that these plants pump oxygen into their roots to oxidize sulfide into the less toxic sulfate[7]. In these environments algaes will preferentially acquire the δ34S of HS- if present rather than the more abundant sulfate, as these sulfides can be readily incorporated into the direct formation of cysteine. This is consistent with cyanobacteria being able to carry out anoxygenic photosynthesis using sulfide[6].
Biochemistry
[edit]~90% of the organic-S in plants is concentrated in the amino acids cysteine and methionine [10]. Cysteine acts as the direct or indirect precursor to any other organic-S compounds in plants such as coenzyme-A, methionine, biotin, lipoic acid and glutathione [7]. The carbon skeleton necessary for S assimilation are provided by glycolysis (acetyl-CoA), respiration (aspartic acid, Asp, which derives from oxaloacetate) and photorespiration (serine, Ser) [2]. Because cysteine is a direct precursor to methionine, methionine is naturally 34S depleted in comparison to cysteine.[2] The majority of sulfur is generally in the organic form but, when excess sulphur is available in the environment, inorganic sulfate becomes the major sulphur form. In most plants, 34S discrimination is minimal and in a study of rice plants it was observed that discrimination takes place in the uptake stage, depleting imported sulfate by 1-2‰ from the source[11]. This effect is through the expression of SO42- transporter genes (SULTR), 14 of which have been identified – which are expressed dependent on the availability of sulfate in the environment. When sulfate is plentiful low affinity transporters are expressed and when sulfate is scarce high affinity genes with greater 34S discrimination [2][11].
Distribution through plant organs
[edit]Sulfate transported through the roots and SO2 diffusing into leaves becomes the pool for plants to assimilate sulfur throughout their tissues. Though there is minimal fractionation from the source sulfur of the total plant organic matter, in wheat, roots and stems are depleted from soil by 2‰ and leaves and grain are 2‰ enriched. The 34S enrichment in leaf whole matter is not caused by 34S-enriched sulfate present in the leaf, but is the result of the 34S-enrichment arriving at sink organs causing proteins in the leaves to be 34S-enriched [2]. In rice, translocation from root to shoot does not discriminate S isotopes, however, the sulfate pools of the shoot are significantly 34S-enriched with respect to the sulfate pools of both root and sap. As sulfate, moves through the plant system and is incorporated into biomass, the pool becomes enriched, giving organs such as leaves and grains a higher δ34S than earlier tissues.[11]
- ^ Grassineau, NV (1998). [chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://rruff.info/doclib/MinMag/Volume_62A/62A-1-537.pdf "Measurement of sulphur isotopic compositions of sulphide minerals using new continuous He-flow EA-MS technology"] (PDF). Mineralogical Magazine. 62: 537–538.
{{cite journal}}: Check|url=value (help); line feed character in|title=at position 32 (help) - ^ a b c d e f Tcherkez, Guillaume; Tea, Illa (2013-10). "32 S/ 34 S isotope fractionation in plant sulphur metabolism". New Phytologist. 200 (1): 44–53. doi:10.1111/nph.12314. ISSN 0028-646X.
{{cite journal}}: Check date values in:|date=(help) - ^ Amrani, Alon; Sessions, Alex L.; Adkins, Jess F. (2009-11-01). "Compound-Specific δ 34 S Analysis of Volatile Organics by Coupled GC/Multicollector-ICPMS". Analytical Chemistry. 81 (21): 9027–9034. doi:10.1021/ac9016538. ISSN 0003-2700.
- ^ Paris, Guillaume; Sessions, Alex L.; Subhas, Adam V.; Adkins, Jess F. (2013-05-08). "MC-ICP-MS measurement of δ34S and ∆33S in small amounts of dissolved sulfate". Chemical Geology. 345: 50–61. doi:10.1016/j.chemgeo.2013.02.022. ISSN 0009-2541.
- ^ Ivanov, M.V. (1981). Some Perspectives of the Major Biogeochemical Cycles. USSR: SCOPE. pp. 61–78.
- ^ a b c d e f Krouse, H. R. (1989). Rundel, P. W.; Ehleringer, J. R.; Nagy, K. A. (eds.). "Sulfur Isotope Studies of the Pedosphere and Biosphere". Stable Isotopes in Ecological Research. New York, NY: Springer: 424–444. doi:10.1007/978-1-4612-3498-2_24. ISBN 978-1-4612-3498-2.
- ^ a b c d Trust, B. A.; Fry, B. (1992-12). "Stable sulphur isotopes in plants: a review". Plant, Cell and Environment. 15 (9): 1105–1110. doi:10.1111/j.1365-3040.1992.tb01661.x. ISSN 0140-7791.
{{cite journal}}: Check date values in:|date=(help) - ^ Wadleigh, Moire A. (2003). "Lichens and atmospheric sulphur: what stable isotopes reveal". Environmental Pollution (Barking, Essex: 1987). 126 (3): 345–351. doi:10.1016/s0269-7491(03)00247-1. ISSN 0269-7491. PMID 12963295.
- ^ a b c De Kok, Luit J.; Durenkamp, Mark; Yang, Liping; Stulen, Ineke (2007), Hawkesford, Malcolm J.; De Kok, Luit J. (eds.), "Atmospheric sulfur", Sulfur in Plants An Ecological Perspective, Dordrecht: Springer Netherlands, pp. 91–106, doi:10.1007/978-1-4020-5887-5_5, ISBN 978-1-4020-5887-5, retrieved 2022-05-27
- ^ a b Tanz, Nicole; Schmidt, Hanns-Ludwig (2010-03-10). "δ 34 S-Value Measurements in Food Origin Assignments and Sulfur Isotope Fractionations in Plants and Animals". Journal of Agricultural and Food Chemistry. 58 (5): 3139–3146. doi:10.1021/jf903251k. ISSN 0021-8561.
- ^ a b c Cavallaro, Viviana; Maghrebi, Moez; Caschetto, Mariachiara; Sacchi, Gian Attilio; Nocito, Fabio Francesco (2022). "Sulfur Stable Isotope Discrimination in Rice: A Sulfur Isotope Mass Balance Study". Frontiers in Plant Science. 13. doi:10.3389/fpls.2022.837517/full. ISSN 1664-462X.
{{cite journal}}: CS1 maint: unflagged free DOI (link)