
H3S28P
H3S28P is an epigenetic modification to the DNA packaging protein histone H3. It is a mark that indicates the phosphorylation the 28th serine residue of the histone H3 protein.
Early mitosis phosphorylation patterns for H3S10 and H3S28 are quite similar, starting at the initiation of chromosomal condensation during prophase. Phosphorylation of H3S28 has transcription effects and acetylates other histones.
Nomenclature
[edit]The name of this modification indicates the protein phosphorylation of serine 28 on histone H3 protein subunit: [1]
| Abbr. | Meaning |
| H3 | H3 family of histones |
| S | standard abbreviation for serine |
| 28 | position of amino acid residue
(counting from N-terminus) |
| P | phosphoryl group |
Serine/threonine/tyrosine phosphorylation
[edit]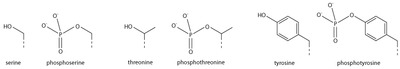
The addition of a negatively charged phosphate group can lead to major changes in protein structure, leading to the well-characterized role of phosphorylation in controlling protein function. It is not clear what structural implications histone phosphorylation has, but histone phosphorylation has clear functions as a post-translational modification.
Effect of modification
[edit]A substantial number of phosphorylated histone residues are associated with gene expression. Interestingly, these are often related to the regulation of proliferative genes. Phosphorylation of serines 10 and 28 of H3 and serine 32 of H2B has been associated with the regulation of epidermal growth factor (EGF)-responsive gene transcription. Depending on the environment in which it happens, the same phosphorylated residue might have drastically different consequences on chromatin structure. H3S10 and H3S28 phosphorylation is an excellent illustration of this duality: both phosphorylated residues are involved in chromatin compaction during mitosis and meiosis, as well as chromatin relaxation after transcription activation.[2] Because of the diverse roles that these phosphorylation events play, individual residue phosphorylation can't be examined in isolation. The impact is determined by the cellular environment and interplay with other cis or trans changes.[2]
Transcription effect
[edit]Early mitosis phosphorylation patterns for H3S10 and H3S28 are quite similar, starting at the initiation of chromosomal condensation during prophase.[3] At gene promoters, H3S28 phosphorylation is expected to remove Polycomb restrictive complexes from chromatin, causing demethylation and acetylation of the nearby K27 residue, so activating transcription. This transition is mediated by the phosphorylation of H3S28 on a site next to H3K27.[3] It was discovered that phosphorylating H3S28 in the promoters of genes like c-fos and -globin controlled their activation.[3]
Acetylation effects
[edit]In addition to H3K27ac, H3S28p plays a role in transcription activation.[3] H3S28p prevents H3K27me3 from accumulating, allowing H3K27 acetylation to occur.[4] Similarly, it was discovered that phosphorylation of H3S28 promotes K9 acetylation.[3]
Specific kinases for modification
[edit]UVB radiation phosphorylates H3S10 and H3S28, while H3S28 is additionally phosphorylated by the kinases MSK1, ERK1, p38, FYN, MLTK, ERK2, and to a lesser extent JNK1 and JNK2.[3]
Histone modifications
[edit]The genomic DNA of eukaryotic cells is wrapped around special protein molecules known as histones. The complexes formed by the looping of the DNA are known as chromatin.
Post-translational modification of histones such as histone phosphorylation has been shown to modify the chromatin structure by changing protein:DNA or protein:protein interactions.[5] Histone post-translational modifications modify the chromatin structure. The most commonly associated histone phosphorylation occurs during cellular responses to DNA damage, when phosphorylated histone H2A separates large chromatin domains around the site of DNA breakage.[6] Researchers investigated whether modifications of histones directly impact RNA polymerase II directed transcription. Researchers choose proteins that are known to modify histones to test their effects on transcription, and found that the stress-induced kinase, MSK1, inhibits RNA synthesis. Inhibition of transcription by MSK1 was most sensitive when the template was in chromatin, since DNA templates not in chromatin were resistant to the effects of MSK1. It was shown that MSK1 phosphorylated histone H2A on serine 1, and mutation of serine 1 to alanine blocked the inhibition of transcription by MSK1. Thus results suggested that the acetylation of histones can stimulate transcription by suppressing an inhibitory phosphorylation by a kinase as MSK1.[7]
Mechanism and function of modification
[edit]Phosphorylation introduces a charged and hydrophilic group in the side chain of amino acids, possibly changing a protein's structure by altering interactions with nearby amino acids. Some proteins such as p53 contain multiple phosphorylation sites, facilitating complex, multi-level regulation. Because of the ease with which proteins can be phosphorylated and dephosphorylated, this type of modification is a flexible mechanism for cells to respond to external signals and environmental conditions.[8]
Kinases phosphorylate proteins and phosphatases dephosphorylate proteins. Many enzymes and receptors are switched "on" or "off" by phosphorylation and dephosphorylation. Reversible phosphorylation results in a conformational change in the structure in many enzymes and receptors, causing them to become activated or deactivated. Phosphorylation usually occurs on serine, threonine, tyrosine and histidine residues in eukaryotic proteins. Histidine phosphorylation of eukaryotic proteins appears to be much more frequent than tyrosine phosphorylation.[9] In prokaryotic proteins phosphorylation occurs on the serine, threonine, tyrosine, histidine or arginine or lysine residues.[9][10] The addition of a phosphate (PO43-) molecule to a non-polar R group of an amino acid residue can turn a hydrophobic portion of a protein into a polar and extremely hydrophilic portion of a molecule. In this way protein dynamics can induce a conformational change in the structure of the protein via long-range allostery with other hydrophobic and hydrophilic residues in the protein.
Epigenetic implications
[edit]The post-translational modification of histone tails by either histone-modifying complexes or chromatin remodeling complexes is interpreted by the cell and leads to complex, combinatorial transcriptional output. It is thought that a histone code dictates the expression of genes by a complex interaction between the histones in a particular region.[11] The current understanding and interpretation of histones comes from two large scale projects: ENCODE and the Epigenomic roadmap.[12] The purpose of the epigenomic study was to investigate epigenetic changes across the entire genome. This led to chromatin states, which define genomic regions by grouping different proteins and/or histone modifications together. Chromatin states were investigated in Drosophila cells by looking at the binding location of proteins in the genome. Use of ChIP-sequencing revealed regions in the genome characterized by different banding.[13] Different developmental stages were profiled in Drosophila as well, an emphasis was placed on histone modification relevance.[14] A look in to the data obtained led to the definition of chromatin states based on histone modifications.[15] Certain modifications were mapped and enrichment was seen to localize in certain genomic regions.
The human genome is annotated with chromatin states. These annotated states can be used as new ways to annotate a genome independently of the underlying genome sequence. This independence from the DNA sequence enforces the epigenetic nature of histone modifications. Chromatin states are also useful in identifying regulatory elements that have no defined sequence, such as enhancers. This additional level of annotation allows for a deeper understanding of cell specific gene regulation.[16][17]
Methods
[edit]The histone mark can be detected in a variety of ways:
1. Chromatin Immunoprecipitation Sequencing (ChIP-sequencing) measures the amount of DNA enrichment once bound to a targeted protein and immunoprecipitated. It results in good optimization and is used in vivo to reveal DNA-protein binding occurring in cells. ChIP-Seq can be used to identify and quantify various DNA fragments for different histone modifications along a genomic region.[18]
2. Micrococcal Nuclease sequencing (MNase-seq) is used to investigate regions that are bound by well-positioned nucleosomes. Use of the micrococcal nuclease enzyme is employed to identify nucleosome positioning. Well-positioned nucleosomes are seen to have enrichment of sequences.[19]
3. Assay for transposase accessible chromatin sequencing (ATAC-seq) is used to look in to regions that are nucleosome free (open chromatin). It uses hyperactive Tn5 transposon to highlight nucleosome localisation.[20][21][22]
References
[edit]- ^ Huang, Suming; Litt, Michael D.; Ann Blakey, C. (30 November 2015). Epigenetic Gene Expression and Regulation. Elsevier Science. pp. 21–38. ISBN 9780127999586.
- ^ a b Komar, Dorota; Juszczynski, Przemyslaw (2020). "Rebelled epigenome: Histone H3S10 phosphorylation and H3S10 kinases in cancer biology and therapy". Clinical Epigenetics. 12 (1): 147. doi:10.1186/s13148-020-00941-2. PMC 7556946. PMID 33054831.
- ^ a b c d e f Rossetto, Dorine; Avvakumov, Nikita; Côté, Jacques (2012). "Histone phosphorylation". Epigenetics. 7 (10): 1098–1108. doi:10.4161/epi.21975. PMC 3469451. PMID 22948226.
- ^ Zhang, Tianyi; Cooper, Sarah; Brockdorff, Neil (2015). "The interplay of histone modifications – writers that read". EMBO Reports. 16 (11): 1467–1481. doi:10.15252/embr.201540945. PMC 4641500. PMID 26474904.
- ^ Sawicka, Anna; Seiser, Christian (1 August 2014). "Sensing core histone phosphorylation – A matter of perfect timing". Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 1839 (8): 711–718. doi:10.1016/j.bbagrm.2014.04.013. PMC 4103482. PMID 24747175.
- ^ Rossetto, Dorine; Avvakumov, Nikita; Côté, Jacques (1 October 2012). "Histone phosphorylation". Epigenetics. 7 (10): 1098–1108. doi:10.4161/epi.21975. ISSN 1559-2294. PMC 3469451. PMID 22948226.
- ^ Zhang, Ye; Griffin, Karen; Mondal, Neelima; Parvin, Jeffrey D. (21 May 2004). "Phosphorylation of Histone H2A Inhibits Transcription on Chromatin Templates". Journal of Biological Chemistry. 279 (21): 21866–21872. doi:10.1074/jbc.M400099200. ISSN 0021-9258. PMID 15010469.
- ^ Johnson LN, Barford D (1993). "The effects of phosphorylation on the structure and function of proteins[J]". Annual Review of Biophysics and Biomolecular Structure. 22 (1): 199–232. doi:10.1146/annurev.bb.22.060193.001215. PMID 8347989.
- ^ a b Ciesla J; Fraczyk T; Rode W (2011). "Phosphorylation of basic amino acid residues in proteins: important but easily missed". Acta Biochim. Pol. 58 (2): 137–47. doi:10.18388/abp.2011_2258. PMID 21623415.
- ^ Deutscher, J.; Saier, J. (2005). "Ser/Thr/Tyr Protein Phosphorylation in Bacteria – for Long Time Neglected, Now Well Established". Journal of Molecular Microbiology and Biotechnology. 9 (3–4): 125–131. doi:10.1159/000089641. PMID 16415586. S2CID 13093867.
- ^ Jenuwein T, Allis CD (August 2001). "Translating the histone code". Science. 293 (5532): 1074–80. doi:10.1126/science.1063127. PMID 11498575. S2CID 1883924.
- ^ Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, et al. (The ENCODE Project Consortium) (June 2007). "Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project". Nature. 447 (7146): 799–816. Bibcode:2007Natur.447..799B. doi:10.1038/nature05874. PMC 2212820. PMID 17571346.
- ^ Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ, van Steensel B (October 2010). "Systematic protein location mapping reveals five principal chromatin types in Drosophila cells". Cell. 143 (2): 212–24. doi:10.1016/j.cell.2010.09.009. PMC 3119929. PMID 20888037.
- ^ Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, et al. (modENCODE Consortium) (December 2010). "Identification of functional elements and regulatory circuits by Drosophila modENCODE". Science. 330 (6012): 1787–97. Bibcode:2010Sci...330.1787R. doi:10.1126/science.1198374. PMC 3192495. PMID 21177974.
- ^ Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, et al. (March 2011). "Comprehensive analysis of the chromatin landscape in Drosophila melanogaster". Nature. 471 (7339): 480–5. Bibcode:2011Natur.471..480K. doi:10.1038/nature09725. PMC 3109908. PMID 21179089.
- ^ Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, et al. (Roadmap Epigenomics Consortium) (February 2015). "Integrative analysis of 111 reference human epigenomes". Nature. 518 (7539): 317–30. Bibcode:2015Natur.518..317.. doi:10.1038/nature14248. PMC 4530010. PMID 25693563.
- ^ Lee, Yun Hwa; Ma, Hui; Tan, Tuan Zea; Ng, Swee Siang; Soong, Richie; Mori, Seiichi; Fu, Xin-Yuan; Zernicka-Goetz, Magdalena; Wu, Qiang (2012). "Protein Arginine Methyltransferase 6 Regulates Embryonic Stem Cell Identity". Stem Cells and Development. 21 (14): 2613–2622. doi:10.1089/scd.2011.0330. PMC 5729635. PMID 22455726.
- ^ "Whole-Genome Chromatin IP Sequencing (ChIP-Seq)" (PDF). Illumina. Archived (PDF) from the original on 3 January 2020. Retrieved 23 October 2019.
- ^ "MAINE-Seq/Mnase-Seq". illumina. Archived from the original on 12 November 2020. Retrieved 23 October 2019.
- ^ Buenrostro, Jason D.; Wu, Beijing; Chang, Howard Y.; Greenleaf, William J. (2015). "ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide". Current Protocols in Molecular Biology. 109: 21.29.1–21.29.9. doi:10.1002/0471142727.mb2129s109. ISBN 9780471142720. PMC 4374986. PMID 25559105.
- ^ Schep, Alicia N.; Buenrostro, Jason D.; Denny, Sarah K.; Schwartz, Katja; Sherlock, Gavin; Greenleaf, William J. (2015). "Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions". Genome Research. 25 (11): 1757–1770. doi:10.1101/gr.192294.115. ISSN 1088-9051. PMC 4617971. PMID 26314830.
- ^ Song, L.; Crawford, G. E. (2010). "DNase-seq: A High-Resolution Technique for Mapping Active Gene Regulatory Elements across the Genome from Mammalian Cells". Cold Spring Harbor Protocols. 2010 (2): pdb.prot5384. doi:10.1101/pdb.prot5384. ISSN 1559-6095. PMC 3627383. PMID 20150147.