
ERCC4






ERCC4 is a protein designated as DNA repair endonuclease XPF that in humans is encoded by the ERCC4 gene. Together with ERCC1, ERCC4 forms the ERCC1-XPF enzyme complex that participates in DNA repair and DNA recombination.[5][6]
The nuclease enzyme ERCC1-XPF cuts specific structures of DNA. Many aspects of these two gene products are described together here because they are partners during DNA repair. The ERCC1-XPF nuclease is an essential activity in the pathway of DNA nucleotide excision repair (NER). The ERCC1-XPF nuclease also functions in pathways to repair double-strand breaks in DNA, and in the repair of "crosslink" damage that harmfully links the two DNA strands.
Cells with disabling mutations in ERCC4 are more sensitive than normal to particular DNA damaging agents, including ultraviolet radiation and to chemicals that cause crosslinking between DNA strands. Genetically engineered mice with disabling mutations in ERCC4 also have defects in DNA repair, accompanied by metabolic stress-induced changes in physiology that result in premature aging.[7] Complete deletion of ERCC4 is incompatible with viability of mice, and no human individuals have been found with complete (homozygous) deletion of ERCC4. Rare individuals in the human population harbor inherited mutations that impair the function of ERCC4. When the normal genes are absent, these mutations can lead to human syndromes, including xeroderma pigmentosum, Cockayne syndrome and Fanconi anemia.
ERCC1 and ERCC4 are the human gene names and Ercc1 and Ercc4 are the analogous mammalian gene names. Similar genes with similar functions are found in all eukaryotic organisms.
Gene
[edit]The human ERCC4 gene can correct the DNA repair defect in specific ultraviolet light (UV)-sensitive mutant cell lines derived from Chinese hamster ovary cells.[8] Multiple independent complementation groups of Chinese hamster ovary (CHO) cells have been isolated,[9] and this gene restored UV resistance to cells of complementation group 4. Reflecting this cross-species genetic complementation method, the gene was called "Excision repair cross-complementing 4"[10]
The human ERCC4 gene encodes the XPF protein of 916 amino acids with a molecular mass of about 104,000 daltons.
Genes similar to ERCC4 with equivalent functions (orthologs) are found in other eukaryotic genomes. Some of the most studied gene orthologs include RAD1 in the budding yeast Saccharomyces cerevisiae, and rad16+ in the fission yeast Schizosaccharomyces pombe.
Protein
[edit]
One ERCC1 molecule and one XPF molecule bind together, forming an ERCC1-XPF heterodimer which is the active nuclease form of the enzyme. In the ERCC1–XPF heterodimer, ERCC1 mediates DNA– and protein–protein interactions. XPF provides the endonuclease active site and is involved in DNA binding and additional protein–protein interactions.[8]
The ERCC4/XPF protein consists of two conserved areas separated by a less conserved region in the middle. The N-terminal area has homology to several conserved domains of DNA helicases belonging to superfamily II, although XPF is not a DNA helicase.[11] The C-terminal region of XPF includes the active site residues for nuclease activity.[12] (Figure 1).
Most of the ERCC1 protein is related at the sequence level to the C terminus of the XPF protein.,[13] but residues in the nuclease domain are not present. A DNA binding "helix-hairpin-helix" domain at the C-terminus of each protein.
By primary sequence and protein structural similarity, the ERCC1-XPF nuclease is a member of a broader family of structure specific DNA nucleases comprising two subunits. Such nucleases include, for example, the MUS81-EME1 nuclease.
Structure-specific nuclease
[edit]
The ERCC1–XPF complex is a structure-specific endonuclease. ERCC1-XPF does not cut DNA that is exclusively single-stranded or double-stranded, but it cleaves the DNA phosphodiester backbone specifically at junctions between double-stranded and single-stranded DNA. It introduces a cut in double-stranded DNA on the 5′ side of such a junction, about two nucleotides away[14] (Figure 2). This structure-specificity was initially demonstrated for RAD10-RAD1, the yeast orthologs of ERCC1 and XPF.[15]
The hydrophobic helix–hairpin–helix motifs in the C-terminal regions of ERCC1 and XPF interact to promote dimerization of the two proteins.[16][17] There is no catalytic activity in the absence of dimerization. Indeed, although the catalytic domain is within XPF and ERCC1 is catalytically inactive, ERCC1 is indispensable for activity of the complex.
Several models have been proposed for binding of ERCC1–XPF to DNA, based on partial structures of relevant protein fragments at atomic resolution.[16] DNA binding mediated by the helix-hairpin-helix domains of ERCC1 and XPF domains positions the heterodimer at the junction between double-stranded and single-stranded DNA.
Nucleotide excision repair (NER)
[edit]During nucleotide excision repair, several protein complexes cooperate to recognize damaged DNA and locally separate the DNA helix for a short distance on either side of the site of a site of DNA damage. The ERCC1–XPF nuclease incises the damaged DNA strand on the 5′ side of the lesion.[14] During NER, the ERCC1 protein interacts with the XPA protein to coordinate DNA and protein binding.
DNA double-strand break (DSB) repair
[edit]Mammalian cells with mutant ERCC1–XPF are moderately more sensitive than normal cells to agents (such as ionizing radiation) that cause double-stranded breaks in DNA.[18][19] Particular pathways of both homologous recombination repair and non-homologous end-joining rely on ERCC1-XPF function.[20][21] The relevant activity of ERCC1–XPF for both types of double-strand break repair is the ability to remove non-homologous 3′ single-stranded tails from DNA ends before rejoining. This activity is needed during a single-strand annealing subpathway of homologous recombination. Trimming of 3’ single-stranded tails is also needed in a mechanistically distinct subpathway of non-homologous end-joining, independent of the Ku proteins[22][19] Homologous integration of DNA, an important technique for genetic manipulation, is dependent on the function of ERCC1-XPF in the host cell.[23]
Interstrand crosslinks repair
[edit]Mammalian cells carrying mutations in ERCC1 or XPF are especially sensitive to agents that cause DNA interstrand crosslinks (ICL)[24] Interstrand crosslinks block the progression of DNA replication, and structures at blocked DNA replication forks provide substrates for cleavage by ERCC1-XPF.[20][25] Incisions may be made on either side of the crosslink on one DNA strand to unhook the crosslink and initiate repair. Alternatively, a double-strand break may be made in the DNA near the ICL, and subsequent homologous recombination repair my involve ERCC1-XPF action. Although not the only nuclease involved, ERCC1–XPF is required for ICL repair during several phases of the cell cycle.[26][27]
Clinical significance
[edit]Xeroderma pigmentosum (XP)
[edit]Some individuals with the rare inherited syndrome xeroderma pigmentosum have mutations in ERCC4. These patients are classified as XP complementation group F (XP-F). Diagnostic features of XP are dry scaly skin, abnormal skin pigmentation in sun-exposed areas and severe photosensitivity, accompanied by a great than 1000-fold increased risk of developing UV radiation-induced skin cancers.[5]
Cockayne syndrome (CS)
[edit]Most XP-F patients show moderate symptoms of XP, but a few show additional symptoms of Cockayne syndrome.[28] Cockayne syndrome (CS) patients exhibit photosensitivity, and also exhibit developmental defects and neurological symptoms.[5][7]
Mutations in the ERCC4 gene can result in the very rare XF-E syndrome.[29] These patients have characteristics of XP and CS, as well as additional neurologic, hepatobiliary, musculoskeletal and hematopoietic symptoms.
Fanconi anemia
[edit]Several human patients with symptoms of Fanconi anemia (FA) have causative mutations in the ERCC4 gene. Fanconi anemia is a complex disease, involving major hematopoietic symptoms. A characteristic feature of FA is the hypersensitivity to agents that cause interstrand DNA crosslinks. FA patients with ERCC4 mutations have been classified as belonging to Fanconi anemia complementation group Q (FANCQ).[28][30]
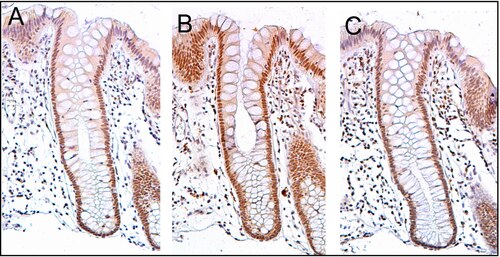
ERCC4 (XPF) is normally expressed at a high level in cell nuclei within the inner surface of the colon (see image, panel C). The inner surface of the colon is lined with simple columnar epithelium with invaginations. The invaginations are called intestinal glands or colon crypts. The colon crypts are shaped like microscopic thick walled test tubes with a central hole down the length of the tube (the crypt lumen). Crypts are about 75 to 110 cells long. DNA repair, involving high expression of ERCC4 (XPF), PMS2 and ERCC1 proteins, appears to be very active in colon crypts in normal, non-neoplastic colon epithelium.
Cells are produced at the crypt base and migrate upward along the crypt axis before being shed into the colonic lumen days later.[32] There are 5 to 6 stem cells at the bases of the crypts.[32] There are about 10 million crypts along the inner surface of the average human colon.[31] If the stem cells at the base of the crypt express ERCC4 (XPF), generally all several thousand cells of the crypt will also express ERCC4 (XPF). This is indicated by the brown color seen by immunostaining of ERCC4 (XPF) in almost all the cells in the crypt in panel C of the image in this section. A similar expression of PMS2 and ERCC1 occurs in the thousands of cells in each normal colonic crypt.
The tissue section in the image shown here was also counterstained with hematoxylin to stain DNA in nuclei a blue-gray color. Nuclei of cells in the lamina propria, cells which are below and surround the epithelial crypts, largely show hematoxylin blue-gray color and have little expression of PMS2, ERCC1 or ERCC4 (XPF). In addition, cells at the very tops of the crypts stained for PMS2 (panel A) or ERCC4 (XPF) (panel C) have low levels of these DNA repair proteins, so that such cells show the blue-gray DNA stain as well.[31]
ERCC4 (XPF) deficiency in the colon epithelium adjacent to and within cancers
[edit]
ERCC4 (XPF) is deficient in about 55% of colon cancers, and in about 40% of the colon crypts in the epithelium within 10 cm adjacent to the cancers (in the field defects from which the cancers likely arose).[31] When ERCC4 (XPF) is reduced in colonic crypts in a field defect, it is most often associated with reduced expression of DNA repair enzymes ERCC1 and PMS2 as well, as illustrated in the image in this section. Deficiencies in ERCC1 (XPF) in colon epithelium appear to be due to epigenetic repression.[31] A deficiency of ERCC4 (XPF) would lead to reduced repair of DNA damages. As indicated by Harper and Elledge,[33] defects in the ability to properly respond to and repair DNA damage underlie many forms of cancer. The frequent epigenetic reduction in ERCC4 (XPF) in field defects surrounding colon cancers as well as in cancers (along with epigenetic reductions in ERCC1 and PMS2) indicate that such reductions may often play a central role in progression to colon cancer.
Although epigenetic reductions in ERCC4 (XPF) expression are frequent in human colon cancers, mutations in ERCC4 (XPF) are rare in humans.[34] However, a mutation in ERCC4 (XPF) causes patients to be prone to skin cancer.[34] An inherited polymorphism in ERCC4 (XPF) appears to be important in breast cancer as well.[35] These infrequent mutational alterations underscore the likely role of ERCC4 (XPF) deficiency in progression to cancer.
Notes
[edit]
The 2015 version of this article was updated by an external expert under a dual publication model. The corresponding academic peer reviewed article was published in Gene and can be cited as: Mandira Manandhar, Karen S Boulware, Richard D. Wood (12 June 2015). "The ERCC1 and ERCC4 (XPF) genes and gene products". Gene. Gene Wiki Review Series. 569 (2): 153–161. doi:10.1016/J.GENE.2015.06.026. ISSN 0378-1119. PMC 4536074. PMID 26074087. Wikidata Q35663361. |
References
[edit]- ^ a b c GRCh38: Ensembl release 89: ENSG00000175595 – Ensembl, May 2017
- ^ a b c GRCm38: Ensembl release 89: ENSMUSG00000022545 – Ensembl, May 2017
- ^ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ a b c Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T (2006). DNA Repair and Mutagenesis. ASM Press. ISBN 978-1555813192.
- ^ "Entrez Gene: ERCC4 excision repair cross-complementing rodent repair deficiency, complementation group 4".
- ^ a b Gregg SQ, Robinson AR, Niedernhofer LJ (Jul 2011). "Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease". DNA Repair. 10 (7): 781–91. doi:10.1016/j.dnarep.2011.04.026. PMC 3139823. PMID 21612988.
- ^ a b Westerveld A, Hoeijmakers JH, van Duin M, de Wit J, Odijk H, Pastink A, Wood RD, Bootsma D (1984). "Molecular cloning of a human DNA repair gene". Nature. 310 (5976): 425–9. Bibcode:1984Natur.310..425W. doi:10.1038/310425a0. PMID 6462228. S2CID 4336902.
- ^ Busch D, Greiner C, Lewis K, Ford R, Adair G, Thompson L (Sep 1989). "Summary of complementation groups of UV-sensitive CHO cell mutants isolated by large-scale screening". Mutagenesis. 4 (5): 349–54. doi:10.1093/mutage/4.5.349. PMID 2687628.
- ^ Brookman KW, Lamerdin JE, Thelen MP, Hwang M, Reardon JT, Sancar A, Zhou ZQ, Walter CA, Parris CN, Thompson LH (Nov 1996). "ERCC4 (XPF) encodes a human nucleotide excision repair protein with eukaryotic recombination homologs". Molecular and Cellular Biology. 16 (11): 6553–62. doi:10.1128/mcb.16.11.6553. PMC 231657. PMID 8887684.
- ^ Sgouros J, Gaillard PH, Wood RD (Mar 1999). "A relationship between a DNA-repair/recombination nuclease family and archaeal helicases". Trends in Biochemical Sciences. 24 (3): 95–7. doi:10.1016/s0968-0004(99)01355-9. PMID 10203755.
- ^ Enzlin JH, Schärer OD (Apr 2002). "The active site of the DNA repair endonuclease XPF-ERCC1 forms a highly conserved nuclease motif". The EMBO Journal. 21 (8): 2045–53. doi:10.1093/emboj/21.8.2045. PMC 125967. PMID 11953324.
- ^ Gaillard PH, Wood RD (Feb 2001). "Activity of individual ERCC1 and XPF subunits in DNA nucleotide excision repair". Nucleic Acids Research. 29 (4): 872–9. doi:10.1093/nar/29.4.872. PMC 29621. PMID 11160918.
- ^ a b Sijbers AM, de Laat WL, Ariza RR, Biggerstaff M, Wei YF, Moggs JG, Carter KC, Shell BK, Evans E, de Jong MC, Rademakers S, de Rooij J, Jaspers NG, Hoeijmakers JH, Wood RD (Sep 1996). "Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease". Cell. 86 (5): 811–22. doi:10.1016/s0092-8674(00)80155-5. hdl:1765/3110. PMID 8797827. S2CID 12957716.
- ^ Bardwell AJ, Bardwell L, Tomkinson AE, Friedberg EC (Sep 1994). "Specific cleavage of model recombination and repair intermediates by the yeast Rad1-Rad10 DNA endonuclease". Science. 265 (5181): 2082–5. Bibcode:1994Sci...265.2082B. doi:10.1126/science.8091230. PMID 8091230.
- ^ a b Tripsianes K, Folkers G, Ab E, Das D, Odijk H, Jaspers NG, Hoeijmakers JH, Kaptein R, Boelens R (December 2005). "The structure of the human ERCC1/XPF interaction domains reveals a complementary role for the two proteins in nucleotide excision repair". Structure. 13 (12): 1849–1858. doi:10.1016/j.str.2005.08.014. hdl:1874/14818. ISSN 1633-8413. PMID 16338413. S2CID 23146316.
- ^ Faridounnia M, Wienk H, Kovačič L, Folkers GE, Jaspers NG, Kaptein R, Hoeijmakers JH, Boelens R (August 2015). "The Cerebro-oculo-facio-skeletal Syndrome Point Mutation F231L in the ERCC1 DNA Repair Protein Causes Dissociation of the ERCC1-XPF Complex". J Biol Chem. 290 (33): 20541–20555. doi:10.1074/jbc.M114.635169. ISSN 1083-351X. PMC 4536458. PMID 26085086.
- ^ Wood RD, Burki HJ, Hughes M, Poley A (Feb 1983). "Radiation-induced lethality and mutation in a repair-deficient CHO cell line". International Journal of Radiation Biology and Related Studies in Physics, Chemistry and Medicine. 43 (2): 207–13. doi:10.1080/09553008314550241. PMID 6600735.
- ^ a b Ahmad A, Robinson AR, Duensing A, van Drunen E, Beverloo HB, Weisberg DB, Hasty P, Hoeijmakers JH, Niedernhofer LJ (Aug 2008). "ERCC1-XPF endonuclease facilitates DNA double-strand break repair". Molecular and Cellular Biology. 28 (16): 5082–92. doi:10.1128/MCB.00293-08. PMC 2519706. PMID 18541667.
- ^ a b Sargent RG, Rolig RL, Kilburn AE, Adair GM, Wilson JH, Nairn RS (Nov 1997). "Recombination-dependent deletion formation in mammalian cells deficient in the nucleotide excision repair gene ERCC1". Proceedings of the National Academy of Sciences of the United States of America. 94 (24): 13122–7. Bibcode:1997PNAS...9413122S. doi:10.1073/pnas.94.24.13122. PMC 24273. PMID 9371810.
- ^ Al-Minawi AZ, Saleh-Gohari N, Helleday T (Jan 2008). "The ERCC1/XPF endonuclease is required for efficient single-strand annealing and gene conversion in mammalian cells". Nucleic Acids Research. 36 (1): 1–9. doi:10.1093/nar/gkm888. PMC 2248766. PMID 17962301.
- ^ Bennardo N, Cheng A, Huang N, Stark JM (Jun 2008). "Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair". PLOS Genetics. 4 (6): e1000110. doi:10.1371/journal.pgen.1000110. PMC 2430616. PMID 18584027.
- ^ Niedernhofer LJ, Essers J, Weeda G, Beverloo B, de Wit J, Muijtjens M, Odijk H, Hoeijmakers JH, Kanaar R (Nov 2001). "The structure-specific endonuclease Ercc1-Xpf is required for targeted gene replacement in embryonic stem cells". The EMBO Journal. 20 (22): 6540–9. doi:10.1093/emboj/20.22.6540. PMC 125716. PMID 11707424.
- ^ Wood RD (Jul 2010). "Mammalian nucleotide excision repair proteins and interstrand crosslink repair". Environmental and Molecular Mutagenesis. 51 (6): 520–6. Bibcode:2010EnvMM..51..520W. doi:10.1002/em.20569. PMC 3017513. PMID 20658645.
- ^ Klein Douwel D, Boonen RA, Long DT, Szypowska AA, Räschle M, Walter JC, Knipscheer P (May 2014). "XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4". Molecular Cell. 54 (3): 460–71. doi:10.1016/j.molcel.2014.03.015. PMC 5067070. PMID 24726325.
- ^ Rahn JJ, Adair GM, Nairn RS (Jul 2010). "Multiple roles of ERCC1-XPF in mammalian interstrand crosslink repair". Environmental and Molecular Mutagenesis. 51 (6): 567–81. Bibcode:2010EnvMM..51..567R. doi:10.1002/em.20583. PMID 20658648. S2CID 29240680.
- ^ Clauson C, Schärer OD, Niedernhofer L (Oct 2013). "Advances in understanding the complex mechanisms of DNA interstrand cross-link repair". Cold Spring Harbor Perspectives in Biology. 5 (10): a012732. doi:10.1101/cshperspect.a012732. PMC 4123742. PMID 24086043.
- ^ a b Kashiyama K, Nakazawa Y, Pilz DT, Guo C, Shimada M, Sasaki K, Fawcett H, Wing JF, Lewin SO, Carr L, Li TS, Yoshiura K, Utani A, Hirano A, Yamashita S, Greenblatt D, Nardo T, Stefanini M, McGibbon D, Sarkany R, Fassihi H, Takahashi Y, Nagayama Y, Mitsutake N, Lehmann AR, Ogi T (May 2013). "Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia". American Journal of Human Genetics. 92 (5): 807–19. doi:10.1016/j.ajhg.2013.04.007. PMC 3644632. PMID 23623389.
- ^ Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst GT, Meinecke P, Kleijer WJ, Vijg J, Jaspers NG, Hoeijmakers JH (Dec 2006). "A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis". Nature. 444 (7122): 1038–43. Bibcode:2006Natur.444.1038N. doi:10.1038/nature05456. PMID 17183314. S2CID 4358515.
- ^ Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, Trujillo JP, Minguillón J, Ramírez MJ, Pujol R, Casado JA, Baños R, Rio P, Knies K, Zúñiga S, Benítez J, Bueren JA, Jaspers NG, Schärer OD, de Winter JP, Schindler D, Surrallés J (May 2013). "Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia". American Journal of Human Genetics. 92 (5): 800–6. doi:10.1016/j.ajhg.2013.04.002. PMC 3644630. PMID 23623386.
- ^ a b c d e f g Facista A, Nguyen H, Lewis C, Prasad AR, Ramsey L, Zaitlin B, Nfonsam V, Krouse RS, Bernstein H, Payne CM, Stern S, Oatman N, Banerjee B, Bernstein C (2012). "Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer". Genome Integr. 3 (1): 3. doi:10.1186/2041-9414-3-3. PMC 3351028. PMID 22494821.
- ^ a b Baker AM, Cereser B, Melton S, Fletcher AG, Rodriguez-Justo M, Tadrous PJ, Humphries A, Elia G, McDonald SA, Wright NA, Simons BD, Jansen M, Graham TA (2014). "Quantification of crypt and stem cell evolution in the normal and neoplastic human colon". Cell Rep. 8 (4): 940–7. doi:10.1016/j.celrep.2014.07.019. PMC 4471679. PMID 25127143.
- ^ Harper JW, Elledge SJ (2007). "The DNA damage response: ten years after". Mol. Cell. 28 (5): 739–45. doi:10.1016/j.molcel.2007.11.015. PMID 18082599.
- ^ a b Gregg SQ, Robinson AR, Niedernhofer LJ (2011). "Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease". DNA Repair (Amst.). 10 (7): 781–91. doi:10.1016/j.dnarep.2011.04.026. PMC 3139823. PMID 21612988.
- ^ Lee E, Levine EA, Franco VI, Allen GO, Gong F, Zhang Y, Hu JJ (2014). "Combined genetic and nutritional risk models of triple negative breast cancer". Nutr Cancer. 66 (6): 955–63. doi:10.1080/01635581.2014.932397. PMID 25023197. S2CID 22065506.
External links
[edit]PDB gallery | |
|---|---|
|



| Excision repair | |
|---|---|
| Homologous recombination | |
| Other pathways | |
| Regulation | |
| Other/ungrouped | |