
Pituitary adenoma
| Pituitary adenoma | |
|---|---|
 | |
| Visual field loss in bitemporal hemianopsia: peripheral vision loss affecting both eyes, resulting from a tumor – typically a pituitary adenoma – putting pressure on the optic chiasm | |
| Specialty | Oncology, endocrinology |
Pituitary adenomas are tumors that occur in the pituitary gland. Most pituitary tumors are benign, approximately 35% are invasive and just 0.1% to 0.2% are carcinomas.[1] Pituitary adenomas represent from 10% to 25% of all intracranial neoplasms, with an estimated prevalence rate in the general population of approximately 17%.[1][2]
Non-invasive and non-secreting pituitary adenomas are considered to be benign in the literal as well as the clinical sense, though a 2011 meta-analysis of available research showed that research to either support or refute this assumption was scant and of questionable quality.[3]
Adenomas exceeding 10 mm (0.39 in) in size are defined as macroadenomas, while those smaller than 10 mm (0.39 in) are referred to as microadenomas. Most pituitary adenomas are microadenomas and have an estimated prevalence of 16.7% (14.4% in autopsy studies and 22.5% in radiologic studies).[2][4] The majority of pituitary microadenomas remain undiagnosed, and those that are diagnosed are often found as an incidental finding and are referred to as incidentalomas.
Pituitary macroadenomas are the most common cause of hypopituitarism.[5][6]
While pituitary adenomas are common, affecting approximately 1 in 6 members of the general population, clinically active pituitary adenomas that require surgical treatment are more rare, affecting approximately 1 in 1,000.[7]
Signs and symptoms
[edit]Physical
[edit]Hormone secreting pituitary adenomas cause one of several forms of hyperpituitarism. The specifics depend on the type of hormone. Some tumors secrete more than one hormone, the most common combination[8] being GH and prolactin, which present as Gigantism or Acromegaly and unexpected lactation (in both men and women).[citation needed]
A patient with pituitary adenoma may present with visual field defects, classically on the left and right in bitemporal hemianopsia. It arises from the compression of the optic nerve by the tumor. The specific area of the visual pathway at which compression by these tumours occurs is at the optic chiasm. If originating superior to the optic chiasm, more commonly in a craniopharyngioma of the pituitary stalk, the visual field defect will first appear as bitemporal inferior quadrantanopia. If originating inferior to the optic chiasm the visual field defect will first appear as bitemporal superior quadrantanopia. Lateral expansion of a pituitary adenoma can also compress the abducens nerve, causing a lateral rectus palsy.[9]
Also, a pituitary adenoma can cause symptoms of increased intracranial pressure. Prolactinomas often start to give symptoms especially during pregnancy, when the increased hormone level estrogen can increase the tumor's growth rate.[10]
Various types of headaches are common in patients with pituitary adenomas. The adenoma may be the prime causative factor behind the headache or may serve to exacerbate a headache caused by other factors. Amongst the types of headaches experienced are both chronic and episodic migraine, and more uncommonly various unilateral headaches; primary stabbing headache,[11] short-lasting unilateral neuralgiform headache attacks with conjunctival injection and tearing (SUNCT)[12] – another type of stabbing headache characterized by short stabs of pain – cluster headache,[13] and hemicrania continua (HS).[14]
Compressive symptoms of pituitary adenomas (visual field deficits, decreased visual acuity, headaches) are more commonly seen with macroadenomas (which are greater than 10 mm in diameter) than with microadenomas (which are less than 10 mm in diameter).[15]
Non-secreting adenomas can go undetected for an extended time because no obvious abnormalities are seen; the gradual reduction in normal activities due to decreased production of hormones is rather less evident. For example, insufficient adrenocorticotropic hormone means that the adrenal glands will not produce sufficient cortisol, resulting in slow recovery from illness, inflammation, and chronic fatigue; insufficient growth hormone in children and adolescents leads to diminished stature but which can have many other explanations.[citation needed]
Psychiatric
[edit]Various psychiatric manifestations have been associated with pituitary disorders including pituitary adenomas. Psychiatric symptoms such as depression, anxiety[16] apathy, emotional instability, easy irritability and hostility have been noted.[17]
Complications
[edit]
- Acromegaly is a syndrome that results when the anterior pituitary gland produces excess growth hormone (GH). Approximately 90–95% of acromegaly cases are caused by a pituitary adenoma and it most commonly affects middle aged adults,[18] Acromegly can result in severe disfigurement, serious complicating conditions, and premature death if unchecked. The disease which is often also associated with gigantism, is difficult to diagnose in the early stages and is frequently missed for many years, until changes in external features, especially of the face, become noticeable with the median time from the development of initial symptoms to diagnosis being twelve years.[19]
- Cushing's syndrome is a hormonal disorder that causes hypercortisolism, which is elevated levels of cortisol in the blood. Cushing's disease (CD) is the most frequent cause of Cushing's syndrome, responsible for approximately 70% of cases.[20] CD results when a pituitary adenoma causes excessive secretion of adrenocorticotropic hormone (ACTH) that stimulates the adrenal glands to produce excessive amounts of cortisol.[21]
- Cushing's disease may cause fatigue, weight gain, fatty deposits around the abdomen and lower back (truncal obesity) and face ("moon face"), stretch marks (striae) on the skin of the abdomen, thighs, breasts, and arms, hypertension, glucose intolerance, and various infections. In women, it may cause excessive growth of facial hair (hirsutism) and in men erectile dysfunction. Psychiatric manifestations may include depression, anxiety, easy irritability, and emotional instability. It may also result in various cognitive difficulties.[citation needed]
- Hyperpituitarism is a disease of the anterior lobe of the pituitary gland which is usually caused by a functional pituitary adenoma and results in hypersecretion of adenohypophyseal hormones such as growth hormone; prolactin; thyrotropin; luteinizing hormone; follicle-stimulating hormone; and adrenocorticotropic hormone.[citation needed]
- Pituitary apoplexy is a condition that occurs when pituitary adenomas suddenly hemorrhage internally, causing a rapid increase in size or when the tumor outgrows its blood supply which causes tissue necrosis and subsequent swelling of the dead tissue. Pituitary apoplexy often presents with visual loss and sudden onset headache and requires timely treatment often with corticosteroids and if necessary surgical intervention.[22]
- Central diabetes insipidus is caused by diminished production of the antidiuretic hormone vasopressin that causes severe thirst and excessive production of very dilute urine (polyuria) which can lead to dehydration. Vasopressin is produced in the hypothalamus and is then transported down the pituitary stalk and stored in the posterior lobe of the pituitary gland which then secretes it into the bloodstream.[23]
As the pituitary gland is in close proximity to the brain, invasive adenomas may invade the dura mater, cranial bone, or sphenoid bone.[24]
Risk factors
[edit]Multiple endocrine neoplasia
[edit]Adenomas of the anterior pituitary gland are a major clinical feature of multiple endocrine neoplasia type 1 (MEN1), a rare inherited endocrine syndrome that affects 1 person in every 30,000. MEN causes various combinations of benign or malignant tumors in various glands in the endocrine system or may cause the glands to become enlarged without forming tumors. It often affects the parathyroid glands, pancreatic islet cells, and anterior lobe of the pituitary gland. MEN1 may also cause non-endocrine tumors such as facial angiofibromas, collagenomas, lipomas, meningiomas, ependymomas, and leiomyomas. Approximately 25 percent of patients with MEN1 develop pituitary adenomas.[25][26]
Carney complex
[edit]Carney complex (CNC), also known as LAMB syndrome[27] and NAME syndrome[27] is an autosomal dominant condition comprising myxomas of the heart and skin, hyperpigmentation of the skin (lentiginosis), and endocrine overactivity and is distinct from Carney's triad.[28][29] Approximately 7% of all cardiac myxomas are associated with Carney complex.[30] Patients with CNC develop growth hormone (GH)-producing pituitary tumors and in some instances these same tumors also secrete prolactin. There are however no isolated prolactinomas or any other type of pituitary tumor. In some patients with CNC, the pituitary gland is characterized by hyperplastic areas with the hyperplasia most likely preceding the formation of GH-producing adenomas.[31]
Familial isolated pituitary adenoma
[edit]Familial isolated pituitary adenoma (FIPA) is a term that is used to identify a condition that displays an autosomal dominant inheritance and is characterised by the presence of two or more related patients affected by adenomas of the pituitary gland only, with no other associated symptoms that occur in multiple endocrine neoplasia type 1 (MEN-1), Carney complex and with mutations in the aryl hydrocarbon receptor-interacting protein (AIP) gene.[32][33][34] FIPA was first described in a limited cohort of families by Albert Beckers group in Liège, Belgium;[35] later FIPA was fully characterized in a multicenter international study of 64 families.[33] FIPA families are divided into those that are homogenous and have the same type of pituitary adenoma in all the affected family members (e.g. only acromegaly, only prolactinoma, etc.), while heterogeneous FIPA families can have different pituitary adenomas in affected family members.[36]
Genetics of FIPA
[edit]FIPA has two known genetic causes, mutations in the AH receptor-interacting protein (AIP) gene[37] and duplications in chromosome Xq26.3 that include the GPR101 gene that also causes X-linked acrogigantism (X-LAG) syndrome.[38] About 15–20% of FIPA families carry a germline AIP gene mutation or deletion, and the disease occurs as autosomal dominant with incomplete penetrance, meaning that about 20% of AIP mutation carriers will develop a pituitary adenoma.[36] AIP mutation associated pituitary adenomas (either presenting as FIPA or as individual, non familial cases) are usually growth hormone-secreting (acromegaly) or prolactin-secreting (prolactinoma) adenomas that are large (macroadenomas) and often occur in children, adolescents and young adults. Daly and colleagues showed that acromegaly cases with AIP mutations occurred about 20 years before acromegaly cases without AIP mutations and these tumors are large and relatively treatment-resistant.[39] Due to their young age at onset, AIP mutations are the most frequent genetic cause of pituitary gigantism (29% of cases).[40]
X-LAG is a rare syndrome of very early childhood onset pituitary tumors/hyperplasia that leads to growth hormone excess and severe overgrowth and pituitary gigantism.[38][41] Three FIPA families with X-LAG have been reported to date all of which had transmission of a chromosome Xq26.3 duplication from affected mother to affected son.[41][38] The disease characteristics of very young onset pituitary gigantism leads to severe overgrowth if not treated adequately; many of the tallest humans in history (e.g. Robert Pershing Wadlow; Sandy Allen, André Rousimoff (Andre the Giant), Zeng Jinlian) had a similar clinical history to patients with X-LAG syndrome.[42] The tallest historical individual with a known genetic cause was Julius Koch (Geant Constantin) who was found to have X-LAG on genetic study of his skeleton.[43] X-LAG has 100% penetrance so far (all affected with the Xq26.3 duplication have the disease and it affects predominantly females.[38] Isolated non familial cases of X-LAG can either have a constitutional duplication of a chromosome Xq26.3 including GPR101, or mosaicism for the duplication (present in a minority of cells) in the case of isolated male patients.[44] X-LAG causes about 10% of cases of pituitary gigantism.[40]
Mechanism
[edit]
The pituitary gland or hypophysis is often referred to as the "master gland" of the human body. Part of the hypothalamic-pituitary axis, it controls most of the body's endocrine functions via the secretion of various hormones into the circulatory system. The pituitary gland is located below the brain in a depression (fossa) of the sphenoid bone known as the sella turcica. Although anatomically and functionally connected to the brain, the pituitary gland[45] sits outside the blood–brain barrier. It is separated from the subarachnoid space by the diaphragma sella, therefore the arachnoid mater and thus cerebral spinal fluid cannot enter the sella turcica.[citation needed]
The pituitary gland is divided into two lobes, the anterior lobe (which accounts for two thirds of the volume of the gland), and the posterior lobe (one third of the volume) separated by the pars intermedia.[8]
The posterior lobe (the neural lobe or neurohypophysis) of the pituitary gland is not, despite its name, a true gland. The posterior lobe contains axons of neurons that extend from the hypothalamus to which it is connected via the pituitary stalk. The hormones vasopressin and oxytocin, produced by the neurons of the supraoptic and paraventricular nuclei of the hypothalamus, are stored in the posterior lobe and released from axon endings (dendrites) within the lobe.[46]
The pituitary gland's anterior lobe (adenohypophysis) is a true gland which produces and secretes six different hormones: thyroid-stimulating hormone (TSH), adrenocorticotropic hormone (ACTH), follicle-stimulating hormone (FSH), luteinizing hormone (LH), growth hormone (GH), and prolactin (PRL).[47]
Diagnosis
[edit]Diagnosis of pituitary adenoma can be made, or at least suspected, by a constellation of related symptoms presented above.[9]
The differential diagnosis includes pituitary tuberculoma, especially in developing countries and in immumocompromised patients.[48] The diagnosis is confirmed by testing hormone levels, and by radiographic imaging of the pituitary (for example, by CT scan or MRI).[citation needed]
Classification
[edit]Pituitary adenomas are classified as endocrine tumors and also as brain tumors, though some patient groups prefer not to refer to them as brain tumors.[49][50][51][52] Pituitary adenomas are classified based upon anatomical, histological and functional criteria.[53]
- Anatomically pituitary tumors are classified by their size based on radiological findings; either microadenomas (less than <10 mm) or macroadenomas (equal or greater than ≥10 mm).
- Classification based on radioanatomical findings places adenomas into 1 of 4 grades (I–IV):[54]
- Stage I: microadenomas (<1 cm) without sella expansion.
- Stage II: macroadenomas (≥1 cm) and may extend above the sella.
- Stage III: macroadenomas with enlargement and invasion of the floor or suprasellar extension.
- Stage IV: destruction of the sella.
- Histological classification utilizes an immunohistological characterization of the tumors in terms of their hormone production.[53] Historically they were classed as either basophilic, acidophilic, or chromophobic on the basis of whether or not they took up the tinctorial stains hematoxylin and eosin. This classification has fallen into disuse, in favor of a classification based on what type of hormone is secreted by the tumor. Approximately 20–25% of adenomas do not secrete any readily identifiable active hormones ('non-functioning tumors') yet they are still sometimes referred to as 'chromophobic'.[citation needed]
- Functional classification is based upon the tumors endocrine activity as determined by serum hormone levels and pituitary tissue cellular hormone secretion detected via immunohistochemical staining.[55] The "Percentage of hormone production cases" values are the fractions of adenomas producing each related hormone of each tumor type as compared to all cases of pituitary tumors, and does not directly correlate to the percentages of each tumor type because of smaller or greater incidences[spelling?] of absence of secretion of the expected hormone. Thus, non secretive adenomas may be either null cell adenomas or a more specific adenoma that, however, remains non-secretive.[citation needed]
- Any type of pituitary adenocarcinoma listed in the table below may cause compressive symptoms due to local expansion in addition to the systemic effects of secreted hormones listed in the pathology column.[citation needed]
- Null cell adenomas by definition do not secrete hormones, but they commonly cause compressive effects on the pituitary stalk (stalk effect). This leads to decreased levels of dopamine from the hypothalamus reaching the anterior pituitary gland. Dopamine exerts an inhibitory effect on prolactin secretion. With the absence of this inhibitory effect, prolactin levels increase and are often increased in null cell adenomas. This leads to symptoms of hypogonadism.[15]
| Type of adenoma | Secretion | Staining | Pathology | Percentage of hormone production cases | Percentage of silent cases[56] |
|---|---|---|---|---|---|
| lactotrophic adenomas (prolactinomas) | secrete prolactin | acidophilic | galactorrhea, hypogonadism, amenorrhea, infertility, and impotence | 30%[57] | <9%[56] |
| somatotrophic adenomas | secrete growth hormone (GH) | acidophilic | acromegaly in adults; gigantism in children | 15%[57] | <9%[56] |
| corticotrophic adenomas | secrete adenocorticotropic hormone (ACTH) | basophilic | Cushing's disease | 2–6%[15] | 10% |
| gonadotrophic adenomas | secrete luteinizing hormone (LH), follicle-stimulating hormone (FSH) and their subunits | basophilic | usually do not cause symptoms, occasionally hypergonadism[15] | 10%[57] | 73%[56] |
| thyrotrophic adenomas (rare) | secrete thyroid-stimulating hormone (TSH) | basophilic to chromophobic | occasionally hyperthyroidism,[58] usually do not cause symptoms | Less than 1%[57] | <9% |
| null cell adenomas | do not secrete hormones | may stain positive for synaptophysin | Asymptomatic or hypogonadism[15] | 25% of pituitary adenomas are nonsecretive[57] | 1%[56] |
-
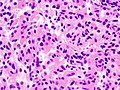 Somatotroph pituitary adenoma, showing acidophilic cytoplasm
Somatotroph pituitary adenoma, showing acidophilic cytoplasm -
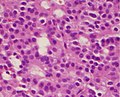 A silent gonadotroph pituitary adenoma which is, in this case, eosinophilic (contrary to normal, basophilic, gonadotroph cells)
A silent gonadotroph pituitary adenoma which is, in this case, eosinophilic (contrary to normal, basophilic, gonadotroph cells) -
![True null cell adenomas are typically composed of uniform, mildly atypical cells with chromophobic cytoplasm. This case has papillary architecture similar to gonadotroph adenomas.[59]](//upload.wikimedia.org/wikipedia/commons/thumb/4/40/Histopathology_of_a_true_null_cell_adenoma.jpg/120px-Histopathology_of_a_true_null_cell_adenoma.jpg) True null cell adenomas are typically composed of uniform, mildly atypical cells with chromophobic cytoplasm. This case has papillary architecture similar to gonadotroph adenomas.[59]
True null cell adenomas are typically composed of uniform, mildly atypical cells with chromophobic cytoplasm. This case has papillary architecture similar to gonadotroph adenomas.[59]
_GH_production.jpg)

![True null cell adenomas are typically composed of uniform, mildly atypical cells with chromophobic cytoplasm. This case has papillary architecture similar to gonadotroph adenomas.[59]](/wiki/File:Histopathology_of_a_true_null_cell_adenoma.jpg)
Pituitary incidentalomas
[edit]Pituitary incidentalomas are pituitary tumors that are characterized as an incidental finding. They are often discovered by computed tomography (CT) or magnetic resonance imaging (MRI), performed in the evaluation of unrelated medical conditions such as suspected head trauma, in cancer staging or in the evaluation of nonspecific symptoms such as dizziness and headache. It is not uncommon for them to be discovered at autopsy. In a meta-analysis, adenomas were found in an average of 16.7% in postmortem studies, with most being microadenomas (<10mm); macrodenomas accounted for only 0.16% to 0.2% of the decedents.[2] While non-secreting, noninvasive pituitary microadenomas are generally considered to be literally as well as clinically benign, a 2011 meta-analysis of available research showed there were, to that time, scant studies of low quality to support this assertion.[3]
The Clinical Practice Guidelines, as published in April 2011 in The Journal of Clinical Endocrinology and Metabolism by the Endocrine Society (a professional, international medical organization), recommend that all patients with pituitary incidentalomas undergo a complete medical history and physical examination, laboratory evaluations to screen for hormone hypersecretion and for hypopituitarism. If the lesion is in close proximity to the optic nerves or optic chiasm, a visual field examination should be performed. For those with incidentalomas which do not require surgical removal, follow up clinical assessments and neuroimaging should be performed as well follow-up visual field examinations for incidentalomas that abut or compress the optic nerve and chiasm and follow-up endocrine testing for macroincidentalomas.[60]
Ectopic pituitary adenoma
[edit]An ectopic (occurring in an abnormal place) pituitary adenoma is a rare type of tumor which occurs outside of the sella turcica, most often in the sphenoid sinus,[61] suprasellar region, nasopharynx and the cavernous sinuses.[62]
Metastases to the pituitary gland
[edit]Carcinomas that metastasize into the pituitary gland are uncommon and typically seen in the elderly,[63][64] with lung and breast cancers being the most prevalent,[65] In breast cancer patients, metastases to the pituitary gland occur in approximately 6–8% of cases.[66]
Symptomatic pituitary metastases account for only 7% of reported cases. In those who are symptomatic diabetes insipidus often occurs, with rates approximately 29–71%. Other commonly reported symptoms include anterior pituitary dysfunction, visual field defects, headache/pain, and ophthalmoplegia.[67]
Treatment
[edit]Treatment options depend on the type of tumor and on its size:
- Prolactinomas (microadenomas and macroadenomas) are most often treated with cabergoline or bromocriptine (both dopamine agonists) as the first line of treatment.[68] Medical treatment usually effectively decreases tumor size as well as alleviates symptoms.[68] Pituitary adenomas are also followed by serial imaging (usually MRI) to detect any increase in size or mass effect on nearby structures. If medical therapy fails, the second option is usually transphenoidal pituitary surgery.[68] A third line therapy is radiation therapy, proton therapy to shrink the tumor.[68]
- Thyrotropinomas are treated with transsphenoidal pituitary surgery as the first line treatment option.[68] Medical therapy is a second line treatment, and thyrotropinomas respond to treatment with somatostatin receptor ligands such as octreotide or lanreotide.[68] In people with thyrotropinomas, treatment with somatostatin receptor ligands normalized thyroid hormone levels in 80-90% of people, and 42% of people had a decreased tumor size.[68]
- Somatotrophic adenomas are primarily treated with transsphenoidal pituitary surgery, especially if symptoms of acromegaly are present.[68] A second line treatment strategy (which can be used if there is a persistent or recurrent mass or symptoms after surgery) utilizes medical therapy, including octreotide or lanreotide, which are long-acting somatostatin analogs. These somatostatin receptor analogs inhibit secretion of growth hormone.[68] They were found to be about 50–55% effective in reducing tumor mass and reducing growth hormone and insulin like growth factor 1 (IGF-1) levels in studies.[15] The growth hormone receptor antagonist pegvisomant is also used in the treatment of somatotrophic adenomas. Pegvisomant blocks the action of growth hormone. It can either be used as monotherapy or combined with a somatostatin analog.[69]
- Corticotropinomas are primarily treated with transsphenoidal pituitary surgery, especially if signs and symptoms of Cushing syndrome are present.[68] Medication therapy is second line, and includes steroidogenesis inhibitors (ketoconazole, metyrapone, osilodrostat, etomidate or mitotane) which decrease production of cortisol, pituitary blockers such as somatostatin receptor ligand pasireotide or the dopamine agonist cabergoline, or the glucocorticoid receptor antagonist mifepristone.[68] These medications can be combined for a synergistic effect. Medication therapy is often used in conjunction with radiation therapy for corticotropinomas.[68]
- Surgery is a common treatment for pituitary tumors. The normal approach is trans-sphenoidal adenectomy, which usually can remove the tumor without affecting the brain or optic nerves.[70]
- Radiation is also used to treat pituitary adenomas. Examples include external beam or proton beam radiation therapy or stereotactic radiosurgery. External radiation of pituitary adenomas can arrest tumor growth for several years but pituitary failure develops within 10 years in most patients necessitating lifelong hormone replacement.[15] Radiation therapy for pituitary adenomas is associated with a four-fold increase in mortality due to cerebrovascular disease.[15] Lifelong monitoring of pituitary hormones is recommended after radiation therapy as hypopituitarism developed in 17% of those undergoing radiation therapy.[68]
See also
[edit]References
[edit]- ^ a b Pituitary Tumors Treatment (PDQ®)–Health Professional Version NIH National Cancer Institute
- ^ a b c Ezzat S, Asa SL, Couldwell WT, Barr CE, Dodge WE, Vance ML, et al. (August 2004). "The prevalence of pituitary adenomas: a systematic review". Cancer. 101 (3): 613–619. doi:10.1002/cncr.20412. PMID 15274075. S2CID 16595581.
- ^ a b Fernández-Balsells MM, Murad MH, Barwise A, Gallegos-Orozco JF, Paul A, Lane MA, et al. (April 2011). "Natural history of nonfunctioning pituitary adenomas and incidentalomas: a systematic review and metaanalysis". The Journal of Clinical Endocrinology and Metabolism. 96 (4): 905–912. doi:10.1210/jc.2010-1054. PMID 21474687.
- ^ Asa SL (August 2008). "Practical pituitary pathology: what does the pathologist need to know?". Archives of Pathology & Laboratory Medicine. 132 (8): 1231–1240. doi:10.5858/2008-132-1231-PPPWDT. PMID 18684022.
- ^ Higham CE, Johannsson G, Shalet SM (November 2016). "Hypopituitarism". Lancet. 388 (10058): 2403–2415. doi:10.1016/S0140-6736(16)30053-8. PMID 27041067. S2CID 208791062.
- ^ Hyperthyroidism unmasked several years after the medical and radiosurgical treatment of an invasive macroprolactinoma inducing hypopituitarism: a case report. L Foppiani, A Ruelle, P Cavazzani, P del Monte – Cases Journal, 2009
- ^ Daly AF, Rixhon M, Adam C, Dempegioti A, Tichomirowa MA, Beckers A (December 2006). "High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium". The Journal of Clinical Endocrinology and Metabolism. 91 (12): 4769–4775. doi:10.1210/jc.2006-1668. PMID 16968795.
- ^ a b Serri O, Somma M, Rasio E, Brazeau P (January 1989). "Growth hormone-releasing factor increases serum prolactin concentrations in normal subjects and in patients with pituitary adenomas". Clinical Endocrinology. 30 (1): 65–75. doi:10.1111/j.1365-2265.1989.tb03728.x. PMID 2505955. S2CID 27780583.
- ^ a b "Acromegaly and Gigantism". The Lecturio Medical Concept Library. Retrieved 26 June 2021.
- ^ Almalki MH, Alzahrani S, Alshahrani F, Alsherbeni S, Almoharib O, Aljohani N, et al. (2015). "Managing Prolactinomas during Pregnancy". Frontiers in Endocrinology. 6: 85. doi:10.3389/fendo.2015.00085. PMC 4443771. PMID 26074878.
- ^ Levy MJ, Matharu MS, Meeran K, Powell M, Goadsby PJ (August 2005). "The clinical characteristics of headache in patients with pituitary tumours". Brain. 128 (Pt 8): 1921–1930. doi:10.1093/brain/awh525. PMID 15888539.
- ^ Matharu MS, Levy MJ, Merry RT, Goadsby PJ (November 2003). "SUNCT syndrome secondary to prolactinoma". Journal of Neurology, Neurosurgery, and Psychiatry. 74 (11): 1590–1592. doi:10.1136/jnnp.74.11.1590. PMC 1738244. PMID 14617728.
- ^ Milos P, Havelius U, Hindfelt B (March 1996). "Clusterlike headache in a patient with a pituitary adenoma. With a review of the literature". Headache. 36 (3): 184–188. doi:10.1046/j.1526-4610.1996.3603184.x. PMID 8984093. S2CID 1681207.
- ^ Levy MJ, Matharu MS, Goadsby PJ (March 2003). "Prolactinomas, dopamine agonists and headache: two case reports". European Journal of Neurology. 10 (2): 169–173. doi:10.1046/j.1468-1331.2003.00549.x. PMID 12603293. S2CID 9475046.
- ^ a b c d e f g h Melmed S (March 2020). "Pituitary-Tumor Endocrinopathies". The New England Journal of Medicine. 382 (10): 937–950. doi:10.1056/NEJMra1810772. PMID 32130815. S2CID 212417223.
- ^ Sievers C, Ising M, Pfister H, Dimopoulou C, Schneider HJ, Roemmler J, et al. (March 2009). "Personality in patients with pituitary adenomas is characterized by increased anxiety-related traits: comparison of 70 acromegalic patients with patients with non-functioning pituitary adenomas and age- and gender-matched controls". European Journal of Endocrinology. 160 (3): 367–373. doi:10.1530/EJE-08-0896. PMID 19073833.
- ^ Weitzner MA, Kanfer S, Booth-Jones M (2005). "Apathy and pituitary disease: it has nothing to do with depression". The Journal of Neuropsychiatry and Clinical Neurosciences. 17 (2): 159–166. doi:10.1176/appi.neuropsych.17.2.159. PMID 15939968.
- ^ "Acromegaly and Gigantism". Merck.com. Retrieved 26 October 2010.
- ^ Nabarro JD (April 1987). "Acromegaly". Clinical Endocrinology. 26 (4): 481–512. doi:10.1111/j.1365-2265.1987.tb00805.x. PMID 3308190. S2CID 221550204.
- ^ Cushing's Syndrome Archived 2011-04-10 at the Wayback Machine at The National Endocrine and Metabolic Diseases Information Service. July 2008. In turn citing: Nieman LK, Ilias I (December 2005). "Evaluation and treatment of Cushing's syndrome". The American Journal of Medicine. 118 (12): 1340–1346. doi:10.1016/j.amjmed.2005.01.059. PMID 16378774.
- ^ Kirk LF, Hash RB, Katner HP, Jones T (September 2000). "Cushing's disease: clinical manifestations and diagnostic evaluation". American Family Physician. 62 (5): 1119–27, 1133–4. PMID 10997535.
- ^ Biousse V, Newman NJ, Oyesiku NM (October 2001). "Precipitating factors in pituitary apoplexy". Journal of Neurology, Neurosurgery, and Psychiatry. 71 (4): 542–545. doi:10.1136/jnnp.71.4.542. PMC 1763528. PMID 11561045.
- ^ Maghnie M (2003). "Diabetes insipidus". Hormone Research. 59 (Suppl 1): 42–54. doi:10.1159/000067844. PMID 12566720. S2CID 24638358.
- ^ Ganapathy MK, Tadi P (2022). "Anatomy, Head and Neck, Pituitary Gland". National Center for Biotechnology Information, U.S. National Library of Medicine. PMID 31855373. Retrieved 26 June 2021.
- ^ Newey PJ, Thakker RV (2011). "Role of multiple endocrine neoplasia type 1 mutational analysis in clinical practice". Endocrine Practice. 17 (Suppl 3): 8–17. doi:10.4158/EP10379.RA. PMID 21454234.
- ^ Marini F, Falchetti A, Luzi E, Tonelli F, Maria Luisa B (2009). "Multiple Endocrine Neoplasia Type 1 (MEN1) Syndrome". In Riegert-Johnson DL (ed.). Cancer Syndromes. National Center for Biotechnology Information (US). PMID 21249756.
- ^ a b Carney Syndrome at eMedicine
- ^ Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL (July 1985). "The complex of myxomas, spotty pigmentation, and endocrine overactivity". Medicine. 64 (4): 270–283. doi:10.1097/00005792-198507000-00007. PMID 4010501. S2CID 20522398.
- ^ McCarthy PM, Piehler JM, Schaff HV, Pluth JR, Orszulak TA, Vidaillet HJ, et al. (March 1986). "The significance of multiple, recurrent, and "complex" cardiac myxomas". The Journal of Thoracic and Cardiovascular Surgery. 91 (3): 389–396. doi:10.1016/S0022-5223(19)36054-4. PMID 3951243.
- ^ Reynen K (December 1995). "Cardiac myxomas". The New England Journal of Medicine. 333 (24): 1610–1617. doi:10.1056/NEJM199512143332407. PMID 7477198.
- ^ Stergiopoulos SG, Abu-Asab MS, Tsokos M, Stratakis CA (2004). "Pituitary pathology in Carney complex patients". Pituitary. 7 (2): 73–82. doi:10.1007/s11102-005-5348-y. PMC 2366887. PMID 15761655.
- ^ Daly AF, Vanbellinghen JF, Beckers A (November 2007). "Characteristics of familial isolated pituitary adenomas". Expert Review of Endocrinology & Metabolism. 2 (6): 725–733. doi:10.1586/17446651.2.6.725. PMID 30290472. S2CID 52924983.
- ^ a b Daly AF, Jaffrain-Rea ML, Ciccarelli A, Valdes-Socin H, Rohmer V, Tamburrano G, et al. (September 2006). "Clinical characterization of familial isolated pituitary adenomas". The Journal of Clinical Endocrinology and Metabolism. 91 (9): 3316–3323. doi:10.1210/jc.2005-2671. PMID 16787992.
- ^ Caimari F, Hernández-Ramírez LC, Dang MN, Gabrovska P, Iacovazzo D, Stals K, et al. (April 2018). "Risk category system to identify pituitary adenoma patients with AIP mutations". Journal of Medical Genetics. 55 (4): 254–260. doi:10.1136/jmedgenet-2017-104957. PMC 5869708. PMID 29440248.
- ^ Valdes-Socin, Hernan & Poncin, J & Stevens, V & Stevenaert, Achille & Beckers, A. (2000). Familial isolated pituitary adenomas unrelated to MEN1 mutations: A follow-up of 27 patients. Ann Endocrinol (Paris). 61.
- ^ a b Beckers A, Aaltonen LA, Daly AF, Karhu A (April 2013). "Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene". Endocrine Reviews. 34 (2): 239–277. doi:10.1210/er.2012-1013. PMC 3610678. PMID 23371967.
- ^ Daly AF, Vanbellinghen JF, Khoo SK, Jaffrain-Rea ML, Naves LA, Guitelman MA, et al. (May 2007). "Aryl hydrocarbon receptor-interacting protein gene mutations in familial isolated pituitary adenomas: analysis in 73 families". The Journal of Clinical Endocrinology and Metabolism. 92 (5): 1891–1896. doi:10.1210/jc.2006-2513. PMID 17244780.
- ^ a b c d Trivellin G, Daly AF, Faucz FR, Yuan B, Rostomyan L, Larco DO, et al. (December 2014). "Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation". The New England Journal of Medicine. 371 (25): 2363–2374. doi:10.1056/nejmoa1408028. PMC 4291174. PMID 25470569.
- ^ Daly AF, Tichomirowa MA, Petrossians P, Heliövaara E, Jaffrain-Rea ML, Barlier A, et al. (November 2010). "Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study". The Journal of Clinical Endocrinology and Metabolism. 95 (11): E373–E383. doi:10.1210/jc.2009-2556. PMID 20685857.
- ^ a b Rostomyan L, Daly AF, Petrossians P, Nachev E, Lila AR, Lecoq AL, et al. (October 2015). "Clinical and genetic characterization of pituitary gigantism: an international collaborative study in 208 patients". Endocrine-Related Cancer. 22 (5): 745–757. doi:10.1530/ERC-15-0320. PMC 6533620. PMID 26187128.
- ^ a b Beckers A, Lodish MB, Trivellin G, Rostomyan L, Lee M, Faucz FR, et al. (June 2015). "X-linked acrogigantism syndrome: clinical profile and therapeutic responses". Endocrine-Related Cancer. 22 (3): 353–367. doi:10.1530/ERC-15-0038. PMC 4433400. PMID 25712922.
- ^ Beckers A, Rostomyan L, Potorac I, Beckers P, Daly AF (June 2017). "X-LAG: How did they grow so tall?". Annales d'Endocrinologie. 78 (2): 131–136. doi:10.1016/j.ando.2017.04.013. hdl:2268/210037. PMID 28457479.
- ^ Beckers A, Fernandes D, Fina F, Novak M, Abati A, Rostomyan L, et al. (February 2017). "Paleogenetic study of ancient DNA suggestive of X-linked acrogigantism". Endocrine-Related Cancer. 24 (2): L17–L20. doi:10.1530/ERC-16-0558. PMID 28049632.
- ^ Daly AF, Yuan B, Fina F, Caberg JH, Trivellin G, Rostomyan L, et al. (April 2016). "Somatic mosaicism underlies X-linked acrogigantism syndrome in sporadic male subjects". Endocrine-Related Cancer. 23 (4): 221–233. doi:10.1530/ERC-16-0082. PMC 4877443. PMID 26935837.
- ^ Dhruve
- ^ Whitehead SA, Nussey SS (2001). Endocrinology: An Integrated Approach. Oxford: BIOS Scientific Publishers. ISBN 978-0-203-45043-7.[page needed]
- ^ Zhao Y, Mailloux CM, Hermesz E, Palkóvits M, Westphal H (January 2010). "A role of the LIM-homeobox gene Lhx2 in the regulation of pituitary development". Developmental Biology. 337 (2): 313–323. doi:10.1016/j.ydbio.2009.11.002. PMC 2832476. PMID 19900438.
- ^ Saini KS, Patel AL, Shaikh WA, Magar LN, Pungaonkar SA (August 2007). "Magnetic resonance spectroscopy in pituitary tuberculoma" (PDF). Singapore Medical Journal. 48 (8): 783–786. PMID 17657390.
- ^ Dai C, Kang J, Liu X, Yao Y, Wang H, Wang R (17 March 2021). "How to Classify and Define Pituitary Tumors: Recent Advances and Current Controversies". Frontiers in Endocrinology. 12: 604644. doi:10.3389/fendo.2021.604644. PMC 8010908. PMID 33815274.
- ^ Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS (October 2021). "CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014-2018". Neuro-Oncology. 23 (12 Suppl 2): iii1–iii105. doi:10.1093/neuonc/noab200. PMC 8491279. PMID 34608945.
- ^ "Benign brain tumour (non-cancerous)". nhs.uk. 20 October 2017. Retrieved 12 August 2024.
- ^ appeal.admin (18 November 2020). "Pituitary tumour vs brain tumour". Pituitary Foundation. Retrieved 12 August 2024.
- ^ a b Ironside JW (August 2003). "Best Practice No 172: pituitary gland pathology". Journal of Clinical Pathology. 56 (8): 561–568. doi:10.1136/jcp.56.8.561. PMC 1770019. PMID 12890801.
- ^ Asa SL, Ezzat S (December 1998). "The cytogenesis and pathogenesis of pituitary adenomas". Endocrine Reviews. 19 (6): 798–827. doi:10.1210/edrv.19.6.0350. PMID 9861546. S2CID 32722411.
- ^ Scanarini M, Mingrino S (1980). "Functional classification of pituitary adenomas". Acta Neurochirurgica. 52 (3–4): 195–202. doi:10.1007/BF01402074. PMID 7424602. S2CID 8095432.
- ^ a b c d e Drummond J, Roncaroli F, Grossman AB, Korbonits M (July 2019). "Clinical and Pathological Aspects of Silent Pituitary Adenomas". The Journal of Clinical Endocrinology and Metabolism. 104 (7): 2473–2489. doi:10.1210/jc.2018-00688. PMC 6517166. PMID 30020466.
- ^ a b c d e Reddy SS, Hamrahian AH (2009). "Pituitary Disorders and Multiple Endocrine Neoplasia Syndromes". In Stoller JK, Michota FA, Mandell BF (eds.). The Cleveland Clinic Foundation Intensive Review of Internal Medicine. Hagerstwon, MD: Lippincott Williams & Wilkins. pp. 525–35. ISBN 978-0-7817-9079-6.
- ^ Chanson P, Weintraub BD, Harris AG (August 1993). "Octreotide therapy for thyroid-stimulating hormone-secreting pituitary adenomas. A follow-up of 52 patients". Annals of Internal Medicine. 119 (3): 236–240. doi:10.7326/0003-4819-119-3-199308010-00010. PMID 8323093. S2CID 27660512.
- ^ Drummond J, Roncaroli F, Grossman AB, Korbonits M (July 2019). "Clinical and Pathological Aspects of Silent Pituitary Adenomas". The Journal of Clinical Endocrinology and Metabolism. 104 (7): 2473–2489. doi:10.1210/jc.2018-00688. PMC 6517166. PMID 30020466.
- "This article has been published under the terms of the Creative Commons Attribution License (CC BY; https://creativecommons.org/licenses/by/4.0/)" - ^ Freda PU, Beckers AM, Katznelson L, Molitch ME, Montori VM, Post KD, et al. (April 2011). "Pituitary incidentaloma: an endocrine society clinical practice guideline". The Journal of Clinical Endocrinology and Metabolism. 96 (4): 894–904. doi:10.1210/jc.2010-1048. PMC 5393422. PMID 21474686.
- ^ Thompson LD, Seethala RR, Müller S (March 2012). "Ectopic sphenoid sinus pituitary adenoma (ESSPA) with normal anterior pituitary gland: a clinicopathologic and immunophenotypic study of 32 cases with a comprehensive review of the english literature". Head and Neck Pathology. 6 (1): 75–100. doi:10.1007/s12105-012-0336-9. PMC 3311955. PMID 22430769.
- ^ Leon Barnes: Pathology and genetics of head and neck tumours; p.100: World Health Organization; (2005) ISBN 92-832-2417-5
- ^ Weil RJ (December 2002). "Pituitary metastasis". Archives of Neurology. 59 (12): 1962–1963. doi:10.1001/archneur.59.12.1962. PMID 12470187.
- ^ Bret P, Jouvet A, Madarassy G, Guyotat J, Trouillas J (May 2001). "Visceral cancer metastasis to pituitary adenoma: report of two cases". Surgical Neurology. 55 (5): 284–290. doi:10.1016/S0090-3019(01)00447-5. PMID 11516470.
- ^ Morita A, Meyer FB, Laws ER (July 1998). "Symptomatic pituitary metastases". Journal of Neurosurgery. 89 (1): 69–73. doi:10.3171/jns.1998.89.1.0069. PMID 9647174.
- ^ Daniel R. Fassett, M.D.; William T. Couldwell, M.D., PhD;Medscape:Metastases to the Pituitary Gland [1]
- ^ Komninos J, Vlassopoulou V, Protopapa D, Korfias S, Kontogeorgos G, Sakas DE, et al. (February 2004). "Tumors metastatic to the pituitary gland: case report and literature review". The Journal of Clinical Endocrinology and Metabolism. 89 (2): 574–580. doi:10.1210/jc.2003-030395. PMID 14764764.
- ^ a b c d e f g h i j k l m Tritos NA, Miller KK (April 2023). "Diagnosis and Management of Pituitary Adenomas: A Review". JAMA. 329 (16): 1386–1398. doi:10.1001/jama.2023.5444. PMID 37097352. S2CID 258310567.
- ^ Neggers SJ, Muhammad A, van der Lely AJ (2016). "Pegvisomant Treatment in Acromegaly". Neuroendocrinology. 103 (1): 59–65. doi:10.1159/000381644. PMID 25792221. S2CID 19588354.
- ^ Transsphenoidal Surgery FAQ Neuroendocrine Clinical Center, Massachusetts General Hospital/Harvard Medical School. Retrieved November 25, 2016